第7章 芳香烃
核心知识点
7.1 苯的结构
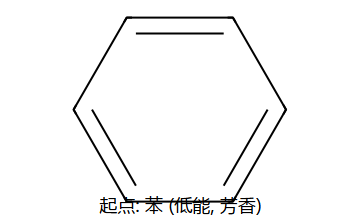
苯的分子式:C6H6,不饱和度 = 4(3个双键 + 1个环)
苯的结构特征
- 6个C均为sp2杂化,每个C有一个未杂化的p轨道
- 所有原子共平面,C-C键长均为0.139 nm(介于单键0.154与双键0.134之间)
- 6个p轨道侧面重叠形成闭合共轭体系
- 键角均为120°,分子具有D6h对称性
共振能(离域能):苯的氢化热 = 208 kJ/mol,比预期的3×环己烯(3×120=360)少152 kJ/mol,说明苯特别稳定。
苯不是"环己三烯":苯不能使Br2/CCl4褪色,不能使KMnO4褪色——与烯烃性质截然不同!苯倾向于发生取代反应而非加成反应,以保持芳香性。
7.2 芳香性与Hückel规则
Hückel规则:判断芳香性的三个条件
- 环状共轭体系(连续共轭)
- 环上所有原子共平面,每个原子都有p轨道参与共轭
- π电子数 = 4n+2(n = 0, 1, 2, ...)→ 2, 6, 10, 14...
| 体系 | π电子数 | 4n+2? | 判断 |
|---|
| 苯 | 6 | n=1 ✓ | 芳香 |
| 环丁二烯 | 4 | 4n(n=1) ✗ | 反芳香 |
| 环辛四烯 | 8 | 4n(n=2) ✗ | 非平面→非芳香 |
| 环戊二烯− | 6 | n=1 ✓ | 芳香 |
| 环庚三烯+ | 6 | n=1 ✓ | 芳香 |
| 环丙烯+ | 2 | n=0 ✓ | 芳香 |
| 环丙烯− | 4 | 4n(n=1) ✗ | 反芳香 |
| [10]轮烯 | 10 | n=2 ✓ | 需共平面才芳香 |
| [14]轮烯 | 14 | n=3 ✓ | 芳香 |
| [18]轮烯 | 18 | n=4 ✓ | 芳香 |
反芳香 vs 非芳香:
• 反芳香:满足环状共轭+共平面,但π电子=4n → 特别不稳定
• 非芳香:不满足环状共轭或不共平面 → 稳定性正常
• 环辛四烯采取船式非平面构象,避免反芳香性,变为非芳香
杂环芳香化合物:吡咯(N提供2个e,6π)、呋喃(O提供2个e,6π)、噻吩(S提供2个e,6π)、吡啶(N提供1个e进入π体系,孤对在sp2中,6π)
Hückel规则的局限性
• 苝(pyrene, 16π电子)、蔻(coronene, 24π电子)虽不符合4n+2但仍具有芳香性
• Hückel规则严格来说不适用于含3个以上稠环的体系(多环稠环芳烃需用其他判据)
• 对于大环轮烯,需同时考虑分子是否能保持平面构象
7.2b 芳烃的物理性质
芳烃的主要物理性质
- 非极性,不溶于水,易溶于有机溶剂;密度d<1(比水轻)
- 对位异构体因分子对称性高,分子间排列紧密,熔点较高(高于邻位和间位异构体)
- 1H NMR:芳环氢的化学位移 δ 6-8 ppm(处于环电流去屏蔽区,是判断芳香性的重要依据)
- 芳烃有特殊气味("芳香"一词的来源),许多芳烃有毒(如苯为致癌物)
7.3 苯的命名
一元取代苯
简单取代基直接命名:甲苯、氯苯、硝基苯、苯酚、苯胺、苯甲酸、苯甲醛等
二元取代苯的位置表示
| 位置关系 | 前缀 | 编号 | 示例 |
|---|
| 1,2- | 邻(ortho-, o-) | 相邻 | 邻二甲苯 |
| 1,3- | 间(meta-, m-) | 间隔一个 | 间二甲苯 |
| 1,4- | 对(para-, p-) | 对面 | 对二甲苯 |
多元取代苯命名:选择取代基编号之和最小的方案;按次序规则列出取代基。
7.4 亲电芳香取代反应(EAS)通用机理
三步机理
① 生成亲电试剂 E+
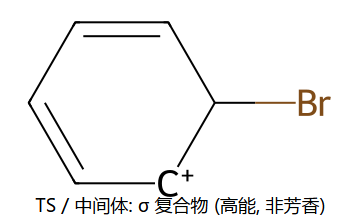
② E+ 进攻苯环π电子 → 形成σ络合物(Wheland中间体,芳香性被破坏)
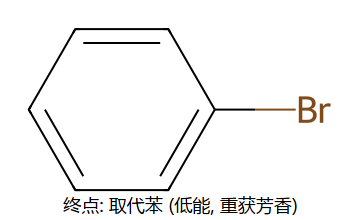
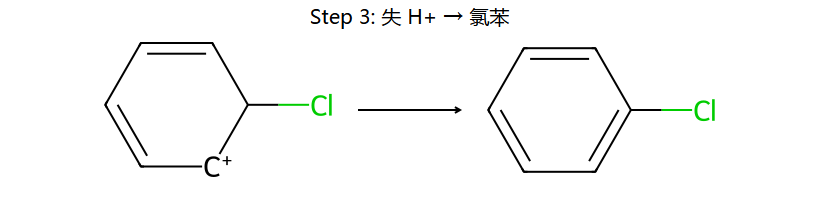
③ σ络合物失去H+ → 恢复芳香性,得到取代产物
为什么取代而不加成? 加成会永久破坏芳香性(损失~152kJ/mol共振能),而取代反应通过失去H+恢复芳香性,热力学更有利。
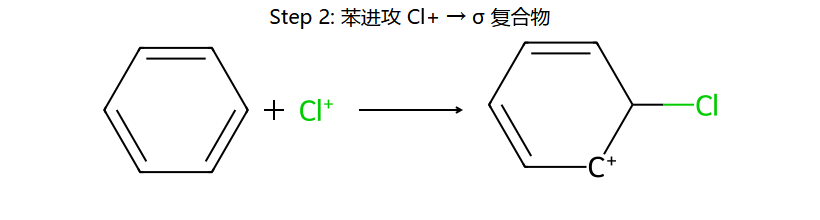
真分子上的 EAS 通用机理 (用 Br+ 代表通用 E+)
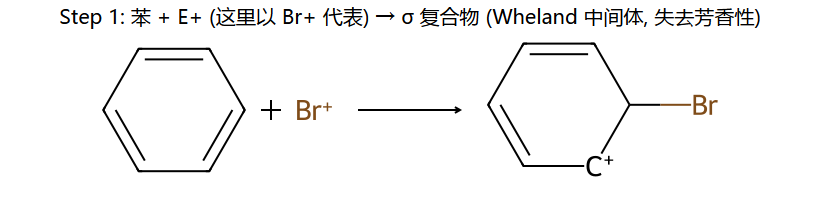
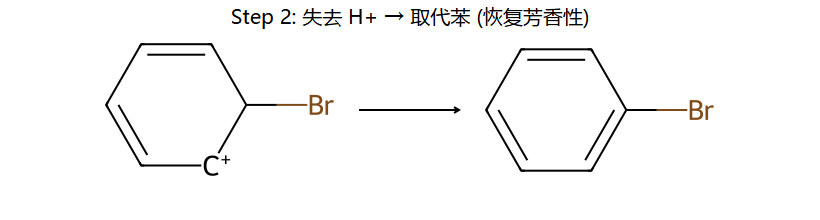
看图重点:Step 1 中苯的芳香性被破坏(中间体的"圈"没了,6π → 4π 离域),所以中间体能量高、能垒大、是决速步。Step 2 一旦失去 H+,芳香性立刻回来(能量大幅下降)→ 整个反应是"暂时牺牲芳香性 → 让 E 加上 → 恢复芳香性"。
7.5 五大亲电取代反应
| 反应 | 试剂/条件 | 亲电试剂E+ | 产物 | 要点 |
|---|
| 硝化 | 浓HNO3/浓H2SO4 | NO2+ | ArNO2 | 混酸,温度控制 |
| 卤代 | Br2(Cl2)/FeBr3(FeCl3) | Br+(Cl+) | ArBr(ArCl) | 必须Lewis酸催化 |
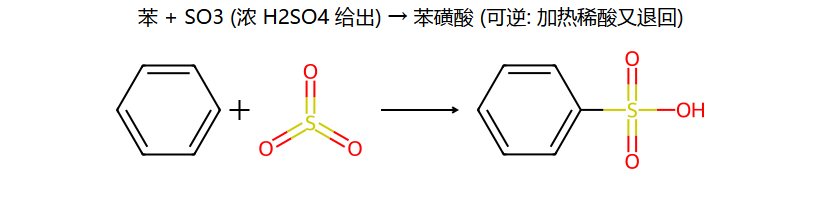
| 磺化 | 发烟H2SO4(含SO3) | SO3 | ArSO3H | 可逆反应! |
| F-C烷基化 | RCl/AlCl3 | R+ | ArR | 会重排+多取代 |
| F-C酰基化 | RCOCl/AlCl3 | RCO+ | ArCOR | 不重排,一元取代 |
① 硝化
亲电试剂的生成
HNO3 + 2H2SO4 → NO2+ + H3O+ + 2HSO4−
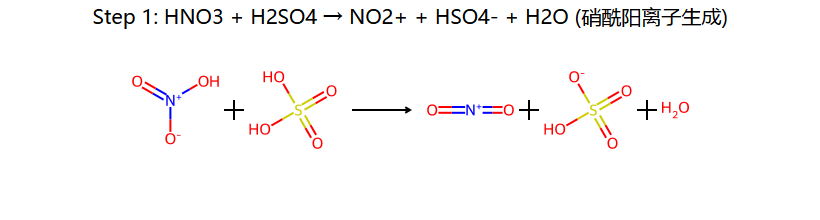
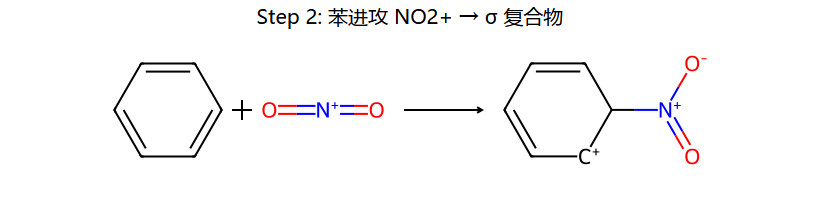
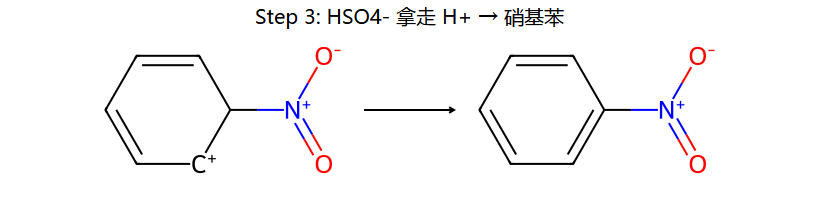
三步串起来:浓 H2SO4 把浓 HNO3 的 OH2+ 踢掉 → 留下线性的 O=N+=O (硝酰阳离子, 极强亲电体);苯进攻它 → σ 复合物;HSO4− 拿走 H+ → 硝基苯。
② 卤代
Lewis酸活化卤素
Br2 + FeBr3 → Br+·FeBr4−(极化产生亲电性Br)
最后:FeBr4− + H+ → FeBr3 + HBr(催化剂再生)
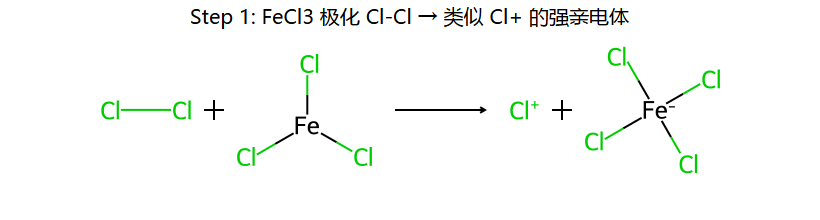
注意区分:Br2/FeBr3 → 苯环上取代;Br2/hv或NBS/hv → 侧链自由基取代(α-H)
③ 磺化
磺化是可逆的! 稀H2SO4水蒸气蒸馏可脱去磺酸基。这使磺酸基可用作"占位基团"——先占位阻挡不想要的位置,后续脱去。
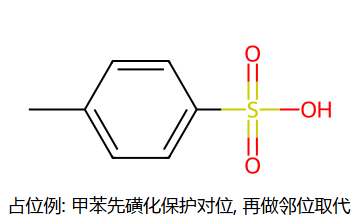
占位法套路:先磺化把位阻好的位置(对位)封掉,后续 EAS 只能进邻位;做完再用稀热酸把 SO3H 蒸掉,邻位产物就纯了。这是合成"邻位选择性 EAS"的常用迂回路线。
④ Friedel-Crafts 烷基化
F-C烷基化的底物类型
- 卤代烃:RCl/AlCl3 → R+(经典底物)
- 烯烃:苯 + CH2=CH2 + AlCl3 → 乙基苯(烯烃质子化产生碳正离子)
- 醇:苯 + ROH + BF3 → 烷基苯(Lewis酸活化醇羟基产生碳正离子)
F-C烷基化的四大问题
- 碳正离子重排:1°卤代烃 → 生成更稳定的2°/3°碳正离子(例:正丙基氯→异丙基苯)
- 多取代:产物ArR比苯更活泼,继续反应生成二取代、三取代
- 不能用于钝化苯环:含-NO2、-COR等吸电子基的苯环不反应
- 不能用于胺:-NH2与AlCl3配位,使催化剂失活
机理实图: 正丙基氯 + 苯/AlCl3 为什么得异丙基苯
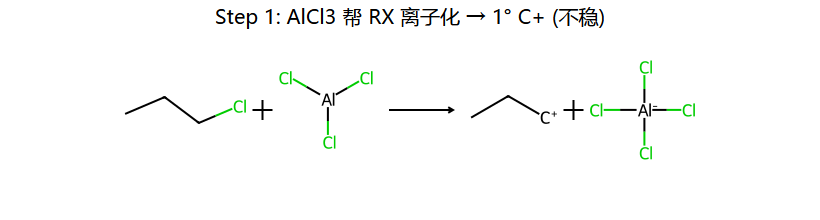
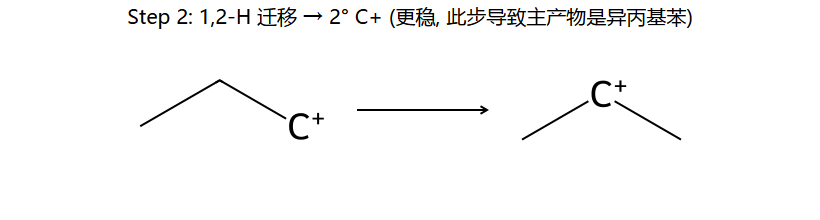
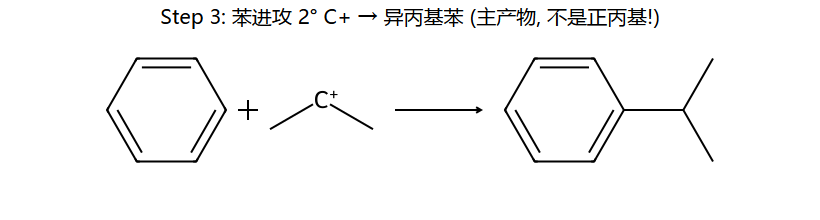
核心陷阱:本想得正丙基苯,但 1° C+ 一旦生成立刻 1,2-H 迁移变成稳定的 2° C+,苯进攻这个 2° C+ → 主产物变成异丙基苯。这是 F-C 烷基化最经典的"重排坑"。
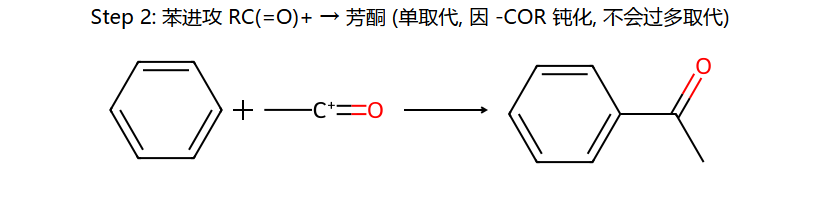
⑤ Friedel-Crafts 酰基化
酰基化的优势
- 不会重排:酰基正离子(RCO+)有共振稳定,不重排
- 只生成一元取代:产物含-COR(吸电子基),使苯环钝化
- 可通过Clemmensen还原(Zn-Hg/HCl)或Wolff-Kishner还原(NH2NH2/KOH)将C=O还原为CH2
- 需要过量AlCl3(>1当量):产物酮的C=O与AlCl3络合,消耗催化剂,因此AlCl3需超过化学计量
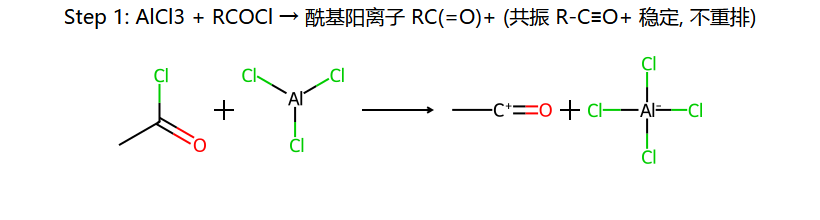
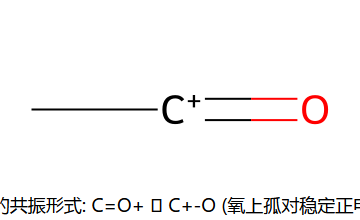
酰基阳离子为什么不重排:C=O+ ↔ C+−O 这对共振让 O 的孤对帮忙稳定正电(实际更像 R−C≡O+),不需要靠骨架重排来求稳。这是它与烷基化(重排乱套)的根本差别。
合成直链烷基苯的策略:避免重排!
苯 →(RCOCl/AlCl3)→ ArCOR →(Zn-Hg/HCl 或 NH2NH2/KOH)→ ArCH2R
酰基化-还原法 = 间接烷基化,避免碳正离子重排
⑥ 氯甲基化反应(Blanc反应)
氯甲基化
苯 + HCHO + HCl,ZnCl2催化,60°C → C6H5CH2Cl(苄氯/氯化苄)
- 机理:甲醛与HCl在Lewis酸催化下形成氯甲基碳正离子中间体 ClCH2+,进攻苯环发生亲电取代
- 产物苄氯是重要的有机合成中间体
- 本质上属于F-C烷基化的一种变体
7.6 定位效应
第一类定位基(邻对位定位基)
活化+邻对位:−NH2, −NHR, −NR2, −OH, −OR, −NHCOR, −OCOCH3, −R, −C6H5
特征:与苯环直接相连的原子上有孤对电子或提供电子的σ键,能通过共振效应(+C)或超共轭向环供电子
特殊:卤素(邻对位+钝化)
−F, −Cl, −Br, −I
吸电子诱导效应(−I) > 供电子共振效应(+C) → 总体钝化
但共振效应决定位置 → 邻对位定位
第二类定位基(间位定位基)
钝化+间位:−NO2, −CF3, −CN, −SO3H, −CHO, −COR, −COOH, −COOR, −CONH2
特征:与苯环直接相连的原子含有多重键(双键或三键连接电负性原子),强吸电子
定位效应的本质:
• 邻对位定位基使邻对位碳上电子密度增大 → E+优先进攻邻对位
• 间位定位基使邻对位碳电子密度降低 → 间位相对最不被钝化
• 实际上间位定位基使所有位置都钝化,但间位钝化最少
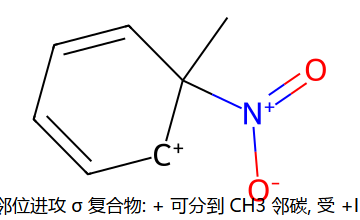
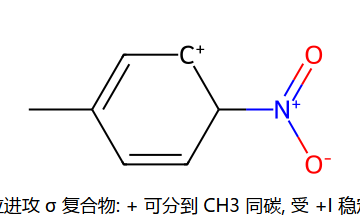
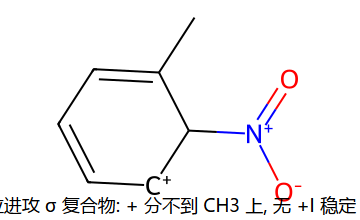
用 σ 复合物共振稳定性解释定位 (甲苯)
看图重点:σ 复合物的正电分布决定了哪条路稳。邻/对位进攻时,正电可以分布到 CH3 邻接的 C 上,受 +I(超共轭+诱导)稳定 → 能垒低 → 主产物。间位进攻时,正电分不到 CH3 上 → 无 +I 稳定 → 能垒高 → 次产物。
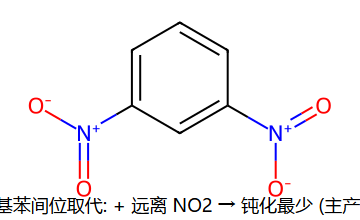
反向:硝基苯 -NO2 是吸电子基,邻/对位进攻时正电会跑到挂 NO2 的 C 上(已经穷的家又被掏空 → 极不稳);间位进攻时正电避开 NO2 → 相对最不糟 → 间位定位 + 整体钝化。
邻对位比例的影响因素
位阻效应:取代基体积越大 → 对位产物比例越高
例:甲苯硝化 → 邻:对 ≈ 58:37;叔丁基苯硝化 → 对位为主(>80%)
7.7 二取代苯定位规则
二取代苯第三个取代基进入位置的判断
- 两个基团定位一致 → 产物明确(协同定位)
- 两个基团定位矛盾 → 以活化能力强的为主
- 一般不进入两个邻位基团之间(位阻太大)
例:对硝基甲苯的溴代,甲基(邻对位)和硝基(间位)矛盾 → 甲基活化更强 → 新基团进入甲基的邻位(同时也是硝基的间位),即2-位。
活化能力排序:−NH2 > −OH > −OR > −NHCOR > −R > −H > 卤素 > −COR > −COOH > −NO2
7.8 苯环侧链反应
三类条件 → 三种反应位置
| 条件 | 反应类型 | 位置 | 示例 |
|---|
| Lewis酸(FeBr3) | 亲电取代 | 苯环上 | 甲苯+Br2/FeBr3→邻/对溴甲苯 |
| hv / NBS / 高温 | 自由基取代 | 侧链α-H | 甲苯+Br2/hv→苄基溴(C6H5CH2Br) |
| KMnO4 | 氧化 | 侧链→COOH | 甲苯→苯甲酸 |
KMnO4氧化侧链的条件:
• 侧链必须有α-H(苄位氢),无α-H不被氧化
• 叔丁基苯(无α-H) → 不反应!
• 无论侧链多长,都氧化为−COOH
• 二甲苯、三甲苯等多侧链 → 多个−COOH
苄基自由基/碳正离子的稳定性:苄基位(α-C)上形成的自由基或碳正离子因与苯环共轭而稳定(类似烯丙基)。NBS(N-溴代丁二酰亚胺)/hv 是苄位溴代的常用试剂。
苯环的催化氧化(苯环破裂)
苯环的工业氧化
苯 + O2,V2O5催化,500°C → 顺丁烯二酸酐(马来酸酐)
- 这是苯环破裂的氧化反应,在工业上用于生产马来酸酐
- 条件剧烈(高温+催化剂),与KMnO4氧化侧链不同
- 注意:实验室条件下苯环对KMnO4稳定,不被氧化
Birch还原
Na/液NH3/ROH(Birch还原)
苯 → 1,4-环己二烯(部分还原,保留两个非共轭双键)
- 供电子基(-OR, -R):供电子基所在碳上保留双键(供电子基连在双键碳上)
- 吸电子基(-COOH):吸电子基所在碳上失去双键(还原发生在吸电子基所在位置)
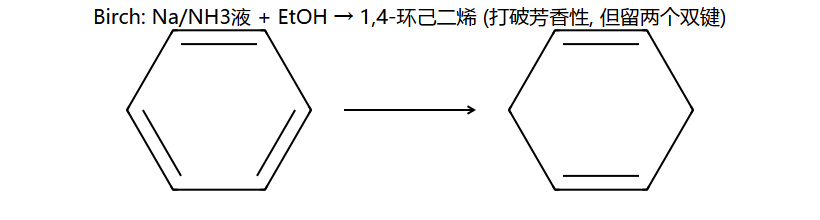
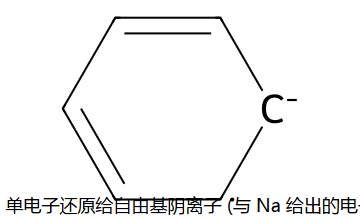
机理简版:Na 在液氨里溶剂化,给出单电子到苯环 → 自由基阴离子;EtOH 给一个 H+,再 Na 给一个电子,再 EtOH 给 H+。共四步轮流,每步只动一对,最终在1,4 位加上 2 个 H → 1,4-环己二烯。"为什么不加 4 个 H 一步到位变环己烷?" — 单电子还原能量分得清楚,不会过头。
苯的加成反应
苯环的加成(需剧烈条件)
- 加氯:苯 + 3Cl2,紫外光照(hv) → C6H6Cl6(六氯环己烷,又称六六六/BHC,自由基加成)
- 催化加氢:苯 + 3H2,Ni催化,175°C,180atm → 环己烷(需高温高压等剧烈条件)
注意:苯的加成需要非常剧烈的条件,正是由于其芳香稳定性。这与苯倾向于发生取代反应(保留芳香性)而非加成反应的规律一致。
7.9 萘的化学
萘的亲电取代
- 萘比苯更容易发生亲电取代(活性更高)
- 动力学控制(低温、温和条件)→ α位取代(过渡态能量更低)
- 热力学控制(高温、剧烈条件)→ β位取代(产物更稳定)
经典考点:萘的磺化
• 低温(80°C)磺化 → α-萘磺酸(动力学产物)
• 高温(160°C)磺化 → β-萘磺酸(热力学产物)
• 这与磺化的可逆性有关:高温下α-产物可脱磺酸再磺化到β位
萘上取代基的定位规则:
• 邻对位定位基 → 新取代基进入同环(优先α位)
• 间位定位基 → 新取代基进入异环(α位)
萘的氧化
萘的氧化反应
- 萘 + CrO3,25°C → 1,4-萘醌(温和条件,部分氧化)
- 萘 + O2,V2O5催化,400-500°C → 邻苯二甲酸酐(工业氧化,萘环完全破裂)
规律:萘环比侧链更容易被氧化(与苯不同!苯环对KMnO4稳定,侧链先被氧化)。萘的芳香性比苯弱,因此萘环本身容易被氧化剂进攻。
萘的还原
萘的还原反应
- 萘 + Na/NH3(液)/乙醇(Birch还原)→ 1,4-二氢萘
- 萘 + H2/Pd → 四氢萘(1,2,3,4-四氢萘,一个环被还原)
- 萘 + H2/高压高温 → 十氢萘(两个环都被完全还原)
7.10 稠环芳烃
常见稠环芳烃
| 名称 | 环数 | π电子数 | 特点 |
|---|
| 萘 | 2 | 10 | 芳香性比苯弱,反应活性更高 |
| 蒽 | 3(线形) | 14 | 9,10-位最活泼 |
| 菲 | 3(角形) | 14 | 9,10-位最活泼 |
• 稠环芳烃的芳香性:萘 > 蒽/菲;环数越多,越容易加成
• 蒽的9,10-位可发生Diels-Alder反应(作为双烯体)
7.11 合成策略总结
常用合成技巧
- 磺酸基占位法:先磺化占位 → 其他反应 → 脱磺酸基
例:合成邻硝基甲苯——甲苯先对位磺化 → 邻位硝化 → 脱磺酸基
- 酰基化-还原法:避免F-C烷基化重排
例:合成正丁基苯——苯先酰基化得ArCOC3H7 → Clemmensen还原
- 先引入定位基:根据目标产物的取代模式,决定引入顺序
例:合成间硝基苯甲酸——先硝化(间位定位) 还是先氧化?
答:先甲基化 → KMnO4氧化 → 再硝化(-COOH间位定位)
- 利用-NH2的转化:-NO2(间位定位) →还原→ -NH2(邻对位定位),灵活切换定位效应
合成题解题步骤:
① 分析目标分子取代基的相对位置(邻/间/对)
② 判断哪个基团先引入(能正确定位后续基团的)
③ 考虑是否需要占位、保护或间接引入
④ 写出完整路线并标注每步试剂和条件
课后习题精选
1. 芳香性判断:判断下列物种的芳香性:(a) 环戊二烯负离子 (b) 环庚三烯正离子 (c) 环辛四烯 (d) 环丙烯正离子 (e) 环丁二烯
答案
(a) 环戊二烯−:5个C各提供1个p轨道,6个π电子(4n+2, n=1),共平面 → 芳香
(b) 环庚三烯+:7个C,6个π电子(4n+2, n=1),共平面 → 芳香
(c) 环辛四烯:8个π电子(4n, n=2),但采取非平面船式构象 → 非芳香(刻意避免反芳香)
(d) 环丙烯+:3个C,2个π电子(4n+2, n=0),共平面 → 芳香
(e) 环丁二烯:4个π电子(4n, n=1),平面 → 反芳香,极不稳定
2. 亲电取代产物:甲苯分别与下列试剂反应,写出主要产物:(a) Br
2/FeBr
3 (b) Br
2/hv (c) HNO
3/H
2SO
4 (d) KMnO
4/H
+
答案
(a) Br2/FeBr3:苯环亲电取代,-CH3为邻对位活化基 → 邻溴甲苯 + 对溴甲苯
(b) Br2/hv:侧链自由基取代,苄位H被取代 → C6H5CH2Br(苄基溴)
(c) HNO3/H2SO4:苯环硝化,-CH3邻对位定位 → 邻硝基甲苯 + 对硝基甲苯
(d) KMnO4/H+:侧链氧化(α-H存在) → 苯甲酸 C6H5COOH
3. 合成题:从苯出发合成
邻硝基苯甲酸
答案
磺酸基占位策略
① 苯 + CH3Cl/AlCl3 → 甲苯
② 甲苯 + 发烟H2SO4 → 对甲苯磺酸(对位被占)
③ 对甲苯磺酸 + HNO3/H2SO4 → 邻位硝化(对位已被磺酸基占据)
④ 稀酸/水蒸气蒸馏 → 脱去磺酸基
⑤ KMnO4 → 氧化甲基为-COOH → 邻硝基苯甲酸
4. 合成题:从苯出发合成
正丁基苯(不能用F-C烷基化直接做,会重排)
答案
酰基化-还原法
① 苯 + CH3CH2CH2COCl/AlCl3 → 苯基丁酮 C6H5COCH2CH2CH3(F-C酰基化,不重排)
② 苯基丁酮 + Zn-Hg/HCl → 正丁基苯(Clemmensen还原)
或 ② 苯基丁酮 + NH2NH2/KOH/Δ → 正丁基苯(Wolff-Kishner还原)
注意:直接用正丁基氯/AlCl3会发生1,2-氢迁移重排,得到仲丁基苯!
5. 定位效应:预测下列化合物硝化的主要产物位置:(a) 苯甲酸 (b) 苯胺 (c) 氯苯 (d) 对硝基甲苯
答案
(a) 苯甲酸:-COOH为间位定位基 → 间硝基苯甲酸
(b) 苯胺:-NH2为强邻对位活化基 → 邻硝基苯胺 + 对硝基苯胺(实际需先保护为-NHCOCH3再硝化,否则-NH2太活泼会被氧化)
(c) 氯苯:-Cl为邻对位定位+钝化 → 邻氯硝基苯 + 对氯硝基苯(反应比苯慢)
(d) 对硝基甲苯:-CH3(邻对位) vs -NO2(间位)矛盾 → -CH3活化更强 → 新基团进入甲基的邻位,即2-硝基-4-硝基甲苯(2,4-二硝基甲苯)
6. 萘的取代:(a) 萘在80°C磺化的主要产物?(b) 在160°C呢?(c) 解释原因。
答案
(a) 80°C → α-萘磺酸(1-萘磺酸),动力学产物
(b) 160°C → β-萘磺酸(2-萘磺酸),热力学产物
(c) 原因:α位取代过渡态能量低,反应速度快(动力学有利)。但β-产物位阻小、更稳定(热力学有利)。磺化可逆,高温下有足够能量使α-产物脱磺酸后重新磺化到β位,达到热力学平衡。
7. 合成题:从甲苯出发合成
对溴苯甲酸
答案
甲苯 + Br2/FeBr3 → 对溴甲苯(-CH3邻对位定位,取对位产物)
对溴甲苯 + KMnO4/H+ → 对溴苯甲酸
注意顺序:不能先氧化!如果先氧化为苯甲酸(-COOH间位定位),再溴代会得到间溴苯甲酸。
8. 合成题:从苯出发合成
间溴硝基苯
答案
分析:Br和NO2处于间位关系。
方案:苯 + HNO3/H2SO4 → 硝基苯 → 硝基苯 + Br2/FeBr3 → 间溴硝基苯
-NO2为间位定位基,Br进入间位。
不能反过来:先溴代再硝化,-Br是邻对位定位,会得到邻/对硝基溴苯。
9. 合成题:从苯出发合成
对硝基苯甲酸
答案
① 苯 + CH3Cl/AlCl3 → 甲苯
② 甲苯 + HNO3/H2SO4 → 对硝基甲苯(-CH3邻对位定位,取对位)
③ 对硝基甲苯 + KMnO4/H+ → 对硝基苯甲酸
关键:先硝化后氧化。不能先氧化(变成-COOH间位定位)再硝化。
10. 比较稳定性:环戊二烯失去H
+的酸性(pK
a≈16)远大于环庚三烯(pK
a≈36),为什么?
答案
环戊二烯失去H+后形成环戊二烯负离子(5个C,6个π电子,满足4n+2规则)→ 芳香稳定
环庚三烯失去H+后形成环庚三烯负离子(7个C,8个π电子,4n)→ 反芳香,极不稳定
因此环戊二烯更容易失去H+,酸性更强。
11. 合成题:从甲苯合成
2,4,6-三硝基甲苯(TNT)
答案
甲苯在混酸(HNO3/H2SO4)中多次硝化:
① 甲苯 + 混酸(稀) → 对硝基甲苯(主产物)+ 邻硝基甲苯
② 对硝基甲苯 + 混酸(较浓) → 2,4-二硝基甲苯
③ 2,4-二硝基甲苯 + 混酸(浓,加热) → 2,4,6-三硝基甲苯(TNT)
-CH3始终为邻对位定位基,三次硝化分别占据2,4,6三个位置。注意每引入一个-NO2会钝化苯环,需要更强条件。
12. 反应条件辨析:写出下列反应的产物
(a) 乙苯 + Cl
2/AlCl
3
(b) 乙苯 + Cl
2/hv
(c) 乙苯 + KMnO
4/H
+加热
答案
(a) 苯环上亲电取代,-C2H5为邻对位定位 → 邻氯乙苯 + 对氯乙苯
(b) 侧链自由基取代,α位C-H键最弱(苄基自由基稳定) → C6H5CHClCH3(1-氯-1-苯基乙烷)
(c) 侧链氧化,有α-H → 苯甲酸 C6H5COOH(不论侧链多长,都变成-COOH)
13. F-C反应:解释为什么用正丙基氯和AlCl
3与苯反应得到的主要产物是
异丙基苯而非正丙基苯?
答案
CH3CH2CH2Cl + AlCl3 → CH3CH2CH2+ (1°碳正离子)
1°碳正离子不稳定,发生1,2-氢迁移 → (CH3)2CH+ (2°碳正离子,更稳定)
2°碳正离子进攻苯环 → 异丙基苯(而非正丙基苯)
这就是F-C烷基化的重排问题。解决方法:用酰基化-还原法。
14. Birch还原:苯甲醚(C
6H
5OCH
3)经Na/液NH
3/EtOH还原的产物是什么?
答案
苯甲醚的-OCH3为供电子基,Birch还原规则:供电子基所在碳保留双键。
产物为2,5-二氢苯甲醚(1-甲氧基-1,4-环己二烯),-OCH3连在双键碳上。
15. 综合合成:从苯和不超过4个碳的有机物出发,合成
对氨基苯甲酸
答案
分析:-NH2(邻对位)和-COOH(间位)在对位关系。
① 苯 + CH3Cl/AlCl3 → 甲苯
② 甲苯 + HNO3/H2SO4 → 对硝基甲苯
③ 对硝基甲苯 + KMnO4 → 对硝基苯甲酸
④ 对硝基苯甲酸 + Fe/HCl(或Sn/HCl) → 对氨基苯甲酸
关键:利用-CH3的邻对位定位引入-NO2到对位,再氧化侧链,最后还原-NO2为-NH2。
历年真题原型(SJTU A 卷实证)
真题原型 1(A 卷 五-1,机理 8 分)——分子内 Friedel-Crafts
PhCH
2CH
2C(CH
3)
2CH
2OH ──H
3PO
4──→ 1-乙基-1-甲基茚满,请画出机理。
完整解法步骤
分子内 F-C 烷基化(醇脱水 + 碳正离子重排 + 关环)
① OH 质子化脱水:H3PO4 把 –CH2OH 质子化成 –CH2OH2+,脱水得到不稳定的 1° 碳正离子 PhCH2CH2C(CH3)2CH2+。
② 1,2-甲基迁移:相邻叔碳上一个 CH3 带电子对迁移到 1° C+ → 生成稳定的 3° 碳正离子 PhCH2CH2C+(CH3)CH2CH3。
③ 分子内亲电进攻苯环(关环):链端的 3° C+ 进攻同分子苯环邻位 C,形成五元碳环融合到苯环上的 σ 络合物(Wheland 中间体)。
④ 失 H+ 重新芳构化 → 得 1-乙基-1-甲基茚满。
得分要点:必须画出每一步电子转移箭头 + 标 C+ 形式电荷 + 写出 1° → 3° 重排的中间体;只写产物拿不到机理分。
真题原型 2(A 卷 六-3,合成 5 分)——磺酸基占位
由苯出发合成
邻硝基苯甲酸。
完整解法步骤
SO3H 先占对位 → 逼硝化到邻位 → 脱磺酸 → 氧化
① 苯 ──CH3Cl/AlCl3──→ 甲苯
② 甲苯 ──浓 H2SO4──→ 对甲苯磺酸(–SO3H 先占阻力小的对位)
③ ──HNO3/H2SO4──→ 对位被磺酸基占据,硝化被逼到邻位(CH3 的邻位)
④ ──稀 H2SO4/H2O, Δ──→ 脱去磺酸基(磺化可逆!)
⑤ ──KMnO4──→ 氧化甲基为 –COOH → 邻硝基苯甲酸
占位逻辑链:先磺化占对位 → 硝化只能去邻位 → 高温脱掉占位的 SO3H → 最后氧化侧链。没有占位步骤就只能得到对硝基产物。
真题原型 3(A 卷 三-1,完成反应)——条件决定环上 vs 侧链
3-溴-4-碘甲苯 ──Cl
2/500℃──→ ? ──CN
−──→ ? ──H
+/H
2O──→ ?
完整解法步骤
① 高温/无 Lewis 酸 = 侧链 α 自由基氯化(不是环上!)→ ArCH2Cl
② ArCH2Cl + CN− ──SN2──→ ArCH2CN
③ ArCH2CN + H+/H2O ──水解──→ ArCH2COOH(芳乙酸)
判别钉子:Cl2/500℃(或 hv)= 侧链;Cl2/FeCl3 = 环上。这是本章最高频的"条件决定位置"陷阱。
📌 必背块(公式·判据·定位序)
芳香性判据(Hückel)
平面 + 环状连续共轭 + 每个环原子有 p 轨道,且 π 电子数 $=4n+2\ (n=0,1,2,\dots)$ → 芳香。
π = $4n$(环状共轭共平面)→ 反芳香(极不稳);不共平面/不连续共轭 → 非芳香。
常考:苯(6π✓)、卓鎓+/环庚三烯+(6π✓)、环戊二烯−(6π✓)、环丙烯+(2π✓);环丁二烯(4π✗反)、环辛四烯(8π,船式非平面→非芳香)。
EAS 五大反应 + 关键标签
硝化(NO2+)|卤化(Br+/FeBr3)|磺化(SO3,可逆 → 占位)|F-C 烷基化(R+,会重排+多取代)|F-C 酰基化(RCO+,不重排+一元)。决速步 = 生成 σ 络合物(芳香性暂时被破坏,能量最高)。
定位效应序(必默写)
邻对位活化:–NR2/–NH2 > –OH > –OCH3 > –NHCOR > –R > –C6H5(注意 –OH 强于 –OCH3)。
间位钝化:–NO2 > –CN > –SO3H > –CHO > –COR > –COOH。
卤素(–F/–Cl/–Br/–I)= 弱钝化但邻对位定位(–I 主导活性、+C 主导位置)。
条件 → 反应位置(铁律)
Lewis 酸(FeBr3/AlCl3) → 环上;hv / NBS / 高温 → 侧链 α;KMnO4(需 α-H)→ 侧链氧化成 –COOH。
萘的取向(动力学/热力学)
低温(80℃)磺化 → α-萘磺酸(动力学);高温(160℃) → β-萘磺酸(热力学,磺化可逆使 α 脱后再上 β)。
⚠️ 易错点
1. F-C 烷基化重排坑:正丙基氯 + 苯/AlCl3 得异丙基苯(1° C+ → 1,2-H 迁移 → 2° C+),不是正丙基苯。要做直链烷基苯必须走酰基化-还原法(RCOCl → ArCOR → Clemmensen/Wolff-Kishner)。酰基化用 RCO+ 共振稳定,不重排。
2. 合成题引入顺序错:要 –COOH 与某基处邻/对位时,先引入 –CH3(邻对位定位)→ 定好位 → 最后 KMnO4 氧化成 –COOH。不能先氧化——一旦变成 –COOH(间位定位)后续基团就跑到间位去了。(对溴苯甲酸、对硝基苯甲酸都踩这个坑)
3. KMnO4 氧化需 α-H:叔丁基苯无 α-H → 不被 KMnO4 氧化。乙基苯/丙基苯不论侧链多长都氧化成 –COOH(只要有 α-H)。
4. "有手性碳 ≠ 有芳香性判断"勿混;芳香性只看 4n+2,反芳香(4n)不是"没芳香"而是更不稳;环辛四烯靠折成船式逃避反芳香 → 归为非芳香。
考前提醒
条件决定位置(必考!)
Lewis酸催化(FeBr3) → 苯环上 | hv/NBS/高温 → 侧链α位 | KMnO4 → 侧链氧化为-COOH
F-C烷基化 vs 酰基化
烷基化:会重排、多取代、可逆、不能用于钝化苯环
酰基化:不重排、一元取代、不可逆
合成题的引入顺序
取代基的相对位置决定引入顺序!邻对位关系 → 先引入邻对位定位基;间位关系 → 先引入间位定位基
磺酸基占位:磺化可逆!利用磺酸基暂时占位,阻挡不需要的位置
KMnO4氧化需α-H:叔丁基苯无α-H不能被KMnO4氧化!
萘:低温α高温β(动力学vs热力学控制)
Birch还原记忆:供电子基"留双键"(双键在取代基碳上),吸电子基"去双键"(取代基碳被还原)