第9章 卤代烃
核心知识点
9.1 分类与命名
按碳骨架分类
| 类型 | 结构 | 示例 |
|---|
| 饱和卤代烃(卤代烷) | R−X | CH3Cl 氯甲烷 |
| 不饱和卤代烃 | 烯丙型 / 乙烯型 | CH2=CHCH2Cl / CH2=CHCl |
| 芳香族卤代烃 | 苄基型 / 苯基型 | C6H5CH2Cl / C6H5Cl |
按卤素所在碳的级别分类
• 伯(1°)卤代烃:R−CH2−X • 仲(2°)卤代烃:R2CH−X • 叔(3°)卤代烃:R3C−X
命名要点:选含卤素的最长碳链为主链;卤素和其他取代基一起按次序规则编号,使位次之和最小;卤素作为取代基(氟、氯、溴、碘)。
9.2 结构与物理性质
C−X键的特征
| 键 | 键长/nm | 键能/(kJ/mol) | 极性 |
|---|
| C−F | 0.140 | 485 | 最强 |
| C−Cl | 0.177 | 339 | 强 |
| C−Br | 0.194 | 285 | 中 |
| C−I | 0.214 | 218 | 弱 |
键能 vs 离去能力:键能 C−F > C−Cl > C−Br > C−I,但离去能力恰好相反:I− > Br− > Cl− > F−。因为离去基团的稳定性(碱性越弱越好离去):HI最强酸 → I−碱性最弱 → 最易离去。
9.3 亲核取代反应 SN2
SN2 机理(双分子亲核取代)
亲核试剂从离去基团的背面进攻,经过五配位过渡态,一步完成:
Nu− + R−X → [Nu···R···X]‡ → Nu−R + X−
SN2 特点
- 一步反应,无中间体,经过过渡态
- 速率 = k[RX][Nu−](双分子,底物和亲核试剂都参与决速步
- Walden翻转(构型反转):S→R 或 R→S
- 活性:CH3X > 1° > 2° >> 3°(位阻越大越不利)
真分子上的 SN2 (用 (S)-2-溴丁烷 + OH−)
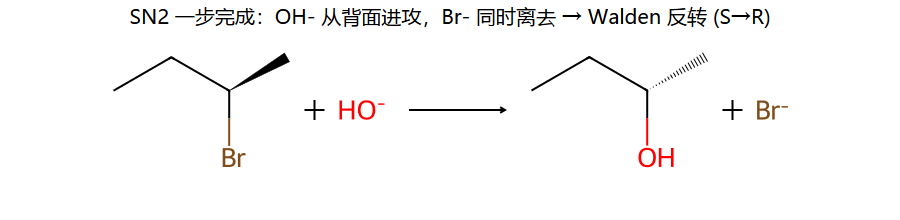
看图重点:左边 (S) 构型的 Br 在前楔形键; 反应后右边 OH 在后楔形键 — 因为 OH⁻ 从 Br 背面进攻, 几何上像伞被风吹翻 (Walden 反转), 构型字母从 S 变 R。
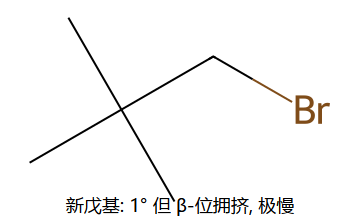
位阻对比:左边 CH3Br 的 C 完全裸露 → 极快;右边新戊基溴虽然是 1°,但 β-C 上三个 CH3 把背面挡死 → 极慢(仅 1° 之一就能慢万倍)。
SN2 不适用于:叔碳(位阻太大)、桥头碳(背面进攻被阻挡)、乙烯基/芳基卤(C=C上卤素不易离去)
分子内SN2反应(成环反应)
分子内亲核取代
分子内的亲核基团攻击同一分子中的碳原子,离去基团离去 → 形成环状产物。
- 五元环最易形成:无角张力,且过渡态中熵因素有利(环化时构象限制适中)
- 三元环(有张力但熵有利)> 四元环(张力大且熵不利)
- 形成难度:5 > 6 > 3 >> 4 >> 大环
经典实例:4-溴-1-丁醇钠的分子内S
N2成环
BrCH2CH2CH2CH2O− → 四氢呋喃(THF) + Br−
氧负离子从分子内部背面进攻C-Br碳,形成五元含氧杂环。
9.4 亲核取代反应 SN1
SN1 机理(单分子亲核取代)
第一步(慢,决速):R−X → R+ + X−(解离生成碳正离子)
第二步(快):R+ + Nu− → R−Nu(亲核试剂进攻碳正离子)
SN1 特点
- 两步反应,经过碳正离子中间体
- 速率 = k[RX](单分子,只有底物参与决速步)
- 外消旋化:碳正离子为平面sp2,Nu可从两面进攻 → 得R+S混合物
- 活性:3° > 2° > 1° > CH3X(碳正离子越稳定越快)
- 可能伴随碳正离子重排(1,2-H迁移或1,2-CH3迁移)
SN1 的重排实例:新戊基溴 (CH3)3CCH2Br 在SN1条件下,1°碳正离子重排为3°碳正离子(1,2-甲基迁移),产物骨架改变。
真分子上的 SN1 机理 2 步 (2-溴-2-甲基丁烷)
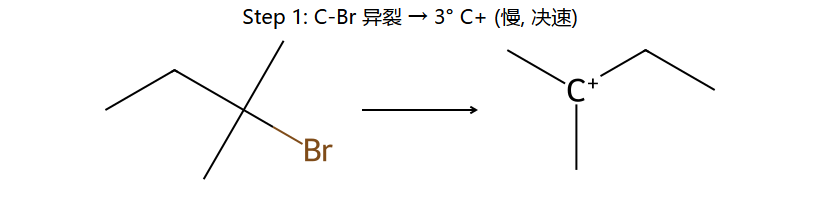
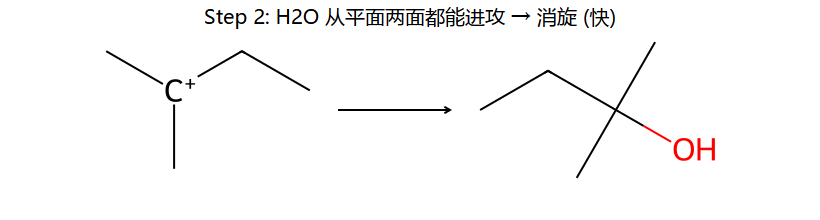
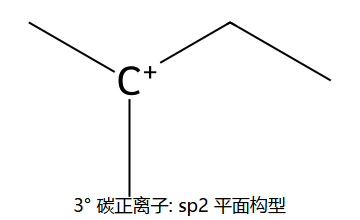
看图重点:Step 1 的决速是 C-Br 自己离了 → 生成平面 sp² 碳正离子(右下小图)。Step 2 因为是平面,H2O 从上面 / 下面进攻概率几乎一样 → 得到一对 R/S 各 50% 的消旋体。这就是 SN1 必定消旋的几何根源。
紧密离子对(Tight Ion Pair)
SN1中的紧密离子对效应
SN1反应中,C−X键异裂后碳正离子与离去基团并不会立即完全分离,而是形成"紧密离子对"(tight/intimate ion pair)。
- 离去基团暂时停留在碳正离子离去的一面 → 部分屏蔽该面
- 亲核试剂从另一面(背面)进攻的概率略高于同面进攻
- 结果:构型翻转略多于保持,而非理想的50:50外消旋化
- 典型比例:约49%构型保持 + 51%构型翻转(不完全外消旋化)
紧密离子对 vs 溶剂分离离子对:随着溶剂化程度增大,离去基团逐渐远离 → 从紧密离子对变为溶剂分离离子对 → 趋向完全外消旋化。
9.5 消除反应
E2 消除(双分子)
E2 机理
强碱夺取β-H,同时C−X键断裂,一步协同完成:
速率 = k[RX][Base]
- 反式共平面(anti-periplanar):H和X必须处于180°反式关系
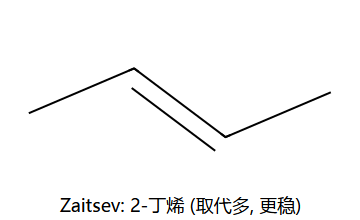
- 产物遵循Saytzeff规则:生成取代最多的烯烃(更稳定)
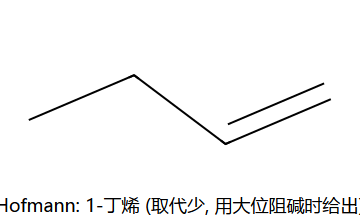
- 若用大位阻碱(如 t-BuOK):得Hofmann产物(取代少的烯烃)
E2反式共平面的推论:
• 环己烷衍生物中,H和X必须处于1,2-双直键(diaxial)位置才能E2消除
• 如果只有一个β-H能满足anti关系 → 只得到一种产物(即使不是Saytzeff产物)
真分子上的 E2 (2-溴丁烷 + OH−)
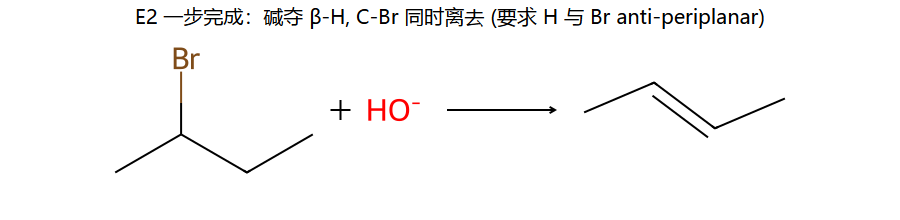
看图重点:碱拿 β-H、Br 同时离开、C=C 同时形成 — 三件事在一个过渡态里同步发生(协同, concerted)。要点:H 和 Br 必须在同一平面、互成 180° (anti-periplanar)。
Saytzeff 主产物 2-丁烯 (取代多, 更稳);Hofmann 次产物 1-丁烯 (用大位阻碱如 t-BuOK 时, 因为碱够不到内位 H, 只能夺取末端 H)。
E1 消除(单分子)
E1 机理
第一步(慢):R−X → R+ + X−(与SN1共享同一步)
第二步(快):碱夺取碳正离子的β-H → 生成烯烃
速率 = k[RX],E1总是伴随SN1发生(竞争关系)
E1 vs E2:E1在SN1条件下伴随发生,无法独立控制;E2可通过强碱诱导,在SN2条件下也可发生。
真分子上的 E1 机理 2 步 (3° 卤代)
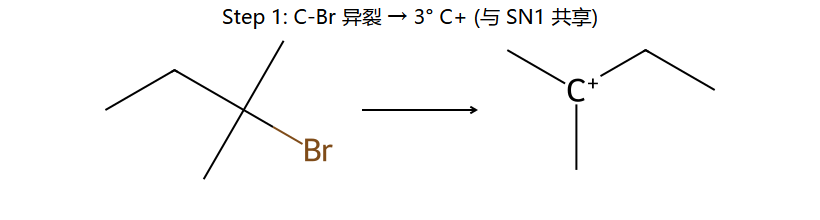
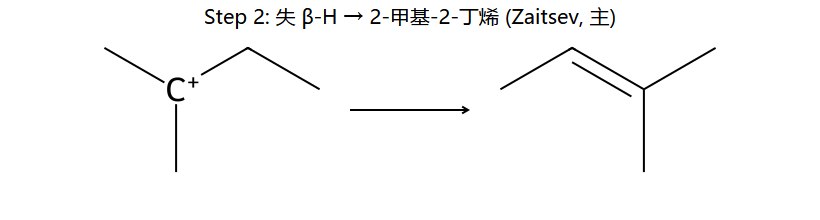
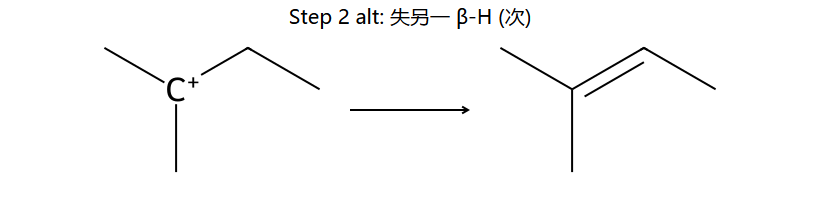
看图重点:Step 1 与 SN1 完全共享(都是 C-Br 自己离)。Step 2 是失 β-H 而非被 Nu 进攻 — 取最多取代的 β 位 = Saytzeff 主产物(左下);少量副产物来自失另一 β 位(右下)。
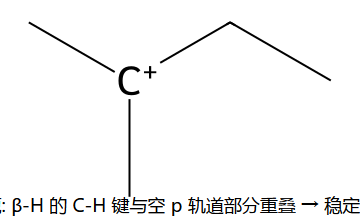
超共轭:3° C⁺ 周围 9 个 β-H 的 σ(C-H) 与空 p 轨道部分重叠 → "电子从 C-H 漏到 C⁺",整体能量下降。烷基越多 → 可参与的 β-H 越多 → 稳定 ↑。这就是 Saytzeff 规则的电子学根源。
超共轭效应解释Saytzeff规则
为什么取代多的烯烃(Saytzeff产物)优先生成?
在E2消除的过渡态中,双键已部分形成:
- 过渡态具有烯烃特征 → 过渡态的稳定性与产物烯烃的稳定性平行(Hammond假说)
- 取代基越多 → 与部分形成的π键发生超共轭(σC-H → π*)越充分 → 过渡态能量越低
- 因此活化能更小 → 反应更快 → 取代最多的烯烃为主要产物
超共轭本质:相邻C−H键的σ电子离域到π体系中,等效于"无键共振"。烷基越多 → 可参与超共轭的C−H键越多 → 稳定化越强。
9.6 四大反应竞争判断(核心考点!)
| SN2 | SN1 | E2 | E1 |
|---|
| 底物 | CH3, 1°最佳 | 3°最佳 | 3° > 2° > 1° | 3° > 2° |
| 试剂 | 强亲核试剂 | 弱亲核/溶剂 | 强碱(尤其大碱) | 弱碱 |
| 溶剂 | 极性非质子 | 极性质子 | 极性非质子/醇 | 极性质子 |
| 温度 | 低温 | — | 高温 | 高温 |
| 动力学 | v=k[RX][Nu] | v=k[RX] | v=k[RX][B] | v=k[RX] |
| 立体化学 | Walden翻转 | 外消旋化 | 反式共平面 | — |
| 重排 | 无 | 有 | 无 | 有 |
快速判断流程
- 看底物:
• CH3X 或 1°RX → SN2(强亲核)或 E2(强碱大碱)
• 3°RX → SN1/E1(弱亲核/溶剂)或 E2(强碱)
• 2°RX → 四种都可能,看试剂和条件
- 看试剂:
• 强亲核弱碱(如 I−, RS−, N3−) → SN2
• 强碱强亲核(如 HO−, RO−) → SN2 或 E2 竞争
• 强碱大位阻(如 t-BuO−, LDA) → E2
• 弱亲核弱碱(如 H2O, ROH) → SN1/E1
- 看溶剂/温度:极性非质子 → 利SN2;极性质子 → 利SN1;高温利消除
弱碱促进取代、抑制消除的技巧
如何获得纯取代产物?
当底物容易发生消除(如仲、叔卤代烃),但希望得到取代产物时:
- 使用弱碱但强亲核试剂:如 CH3COO−(醋酸根)、I−、N3−、RS−
- 弱碱性 → 不易夺取β-H → 抑制消除
- 强亲核性 → 有效进攻碳中心 → 促进取代
间接法制醇(避免直接用强碱HO−导致消除):
- RX + CH3COONa → ROOCCH3(酯,SN2取代)
- ROOCCH3 + NaOH/H2O → ROH + CH3COONa(水解得醇)
溶剂极性对取代/消除竞争的影响
溶剂极性与反应类型选择
- 溶剂极性增大 → 有利于取代反应:取代过渡态中电荷更集中(亲核试剂接近碳中心),极性溶剂稳定此过渡态
- 溶剂极性增大 → 不利于消除反应:消除过渡态中电荷较分散(碱夺H,同时C-X断裂,电荷分布在较大范围内),极性溶剂无法有效稳定
实用规律:
| 条件 | 主要产物 | 原因 |
|---|
| KOH/醇溶液 | 消除为主 | 醇溶剂极性较低,碱浓度高 |
| KOH/水溶液 | 取代为主 | 水极性更大,稳定取代过渡态 |
记忆口诀:醇中消除水中代——KOH/醇 → 消除;KOH/水 → 取代。
9.7 亲核试剂与离去基团
离去基团能力排序
−OTs ≈ −OMs > I− > Br− > Cl− > F− >> HO−, RO−, H2N−
规律:共轭酸越强 → 碱性越弱 → 越好离去
亲核性排序
碱性与亲核性不完全相同!
- 同族元素(质子溶剂中):I− > Br− > Cl− > F−(可极化性主导)
- 同周期元素:碱性越强,亲核性越强。如 NH2− > HO− > F−
- 负离子 > 对应中性分子:HO− > H2O;RO− > ROH
碱性 vs 亲核性:
• 碱性:与H+结合的热力学能力(用pKa衡量)
• 亲核性:与碳原子成键的动力学能力(受可极化性、位阻影响)
• 例如:I−碱性弱但亲核性很强(大原子可极化);t-BuO−碱性强但亲核性差(位阻大)
9.8 溶剂效应
溶剂对反应类型的影响
| 溶剂类型 | 示例 | 特征 | 有利反应 |
|---|
| 极性质子溶剂 | H2O, ROH, RCOOH | 能形成氢键,稳定离子 | SN1 / E1 |
| 极性非质子溶剂 | DMSO, DMF, 丙酮 | 极性强但无活泼H | SN2 |
为什么?
• 极性质子溶剂通过氢键溶剂化亲核试剂,降低其亲核性 → 不利SN2
• 极性质子溶剂稳定碳正离子中间体 → 有利SN1
• 极性非质子溶剂不与阴离子形成氢键 → 亲核试剂保持"裸露"高活性 → 有利SN2
9.9 卤代烃的活性规律
不同类型卤代烃的反应活性
| 类型 | SN活性 | 原因 |
|---|
| 烯丙基型 CH2=CHCH2X | 很活泼(SN1和SN2都快) | SN1:烯丙基碳正离子共振稳定
SN2:p轨道协助背面进攻 |
| 苄基型 ArCH2X | 很活泼 | 同烯丙基,苄基碳正离子与苯环共振 |
| 普通烷基 RX | 正常 | 按1°/2°/3°规律 |
| 乙烯基型 CH2=CHX | 极惰性 | C=C的p轨道与C-X重叠 → 部分双键特征 → 不易断裂 |
| 芳基型 ArX | 极惰性 | 同乙烯基,苯环p轨道与C-X共轭 |
| 桥头碳卤代物 | 不反应 | SN1:桥头无法形成平面碳正离子
SN2:桥头背面被阻挡 |
活性顺序记忆:烯丙基/苄基 >> 3° > 2° > 1° > CH3X >> 乙烯基/芳基/桥头(几乎不反应)
9.10 卤代烃的制备
卤代烃的主要制备方法
- 烷烃卤代(自由基取代):RH + X2 →(hv)→ RX + HX
- 烯烃加HX:遵循Markovnikov规则;过氧化物存在时反Markovnikov(仅HBr)
- 醇 + HX:ROH + HX → RX + H2O(SN1或SN2)
- 醇 + SOCl2:ROH + SOCl2 → RCl + SO2 + HCl(无吡啶:构型保持,SNi机理;有吡啶:构型翻转,SN2机理)
- 醇 + PBr3:3ROH + PBr3 → 3RBr + H3PO3
- 卤素交换(Finkelstein):RCl + NaI →(丙酮)→ RI + NaCl↓(利用NaCl在丙酮中不溶)
- 烯丙位/苄位溴代:NBS/hv → 烯丙位或苄位溴代的专用试剂
- 醇 + PCl3:3ROH + PCl3 → 3RCl + H3PO3
- 醇 + PCl5:ROH + PCl5 → RCl + POCl3 + HCl
Zn脱卤还原制备烯烃
邻二卤代物与锌粉反应,脱去两个卤素生成烯烃:
RCHBr−CHBrR' + Zn → RCH=CHR' + ZnBr2
- 反应条件:Zn粉/醇或Zn粉/醋酸
- 本质是还原消除(脱卤化氢的逆反应为加卤;此为脱卤)
- 应用:烯烃 →(Br2加成)→ 邻二溴代物(用于保护双键)→ Zn脱卤还原恢复双键
9.11 Grignard试剂与有机金属化合物
格氏试剂 RMgX
RX + Mg →(无水乙醚)→ RMgX(格氏试剂)
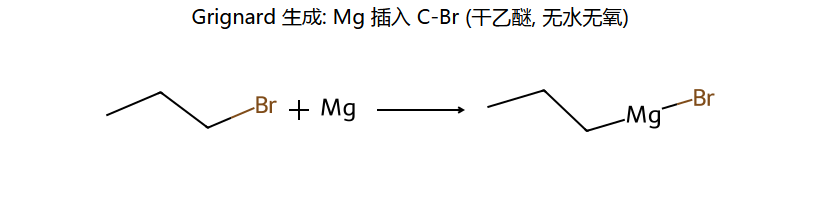
看图重点:Mg 把自己插进 C-Br 键里,碳上原来有的部分负电荷被强化(因为 Mg 是电正性金属),所以 RMgX 中的 R 是强亲核体 / 强碱。这是格氏试剂能做几乎所有"加 C"反应的本质。
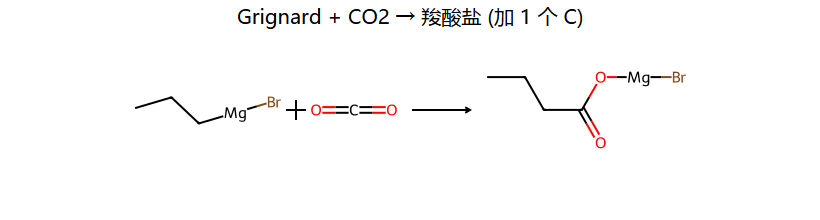
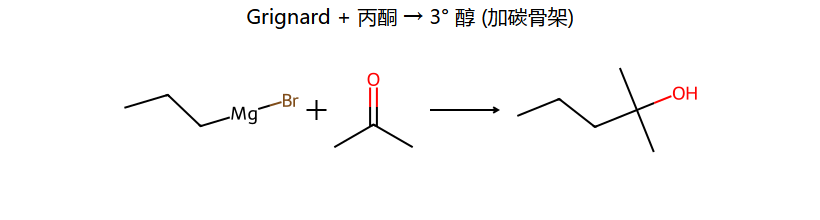
三大经典反应(看图记)
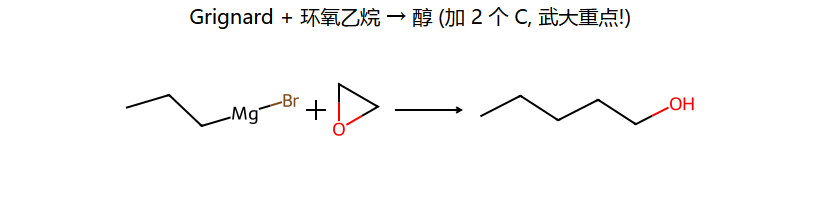
武大第六版重点:环氧乙烷 + RMgBr → 醇增 2 个 C — 这是醛/酮"增 1 C"之外的另一条增碳路线,频繁出现在合成题里。
| 反应物 | 产物 | 用途 |
|---|
| RMgX + H2O(D2O) | RH (RD) | 引入氘标记 |
| RMgX + CO2 | RCOOH | 增1C得羧酸 |
| RMgX + 环氧乙烷 | RCH2CH2OH | 增2C得伯醇 |
| RMgX + HCHO | RCH2OH | 增1C得伯醇 |
| RMgX + RCHO | R2CHOH | 得仲醇 |
| RMgX + R2CO | R3COH | 得叔醇 |
| RMgX + RX' | R−R' | 偶联(产率低) |
格氏试剂不能与活泼氢共存!
−OH, −NH, −SH, −COOH, −C≡CH 都含活泼氢,会与RMgX反应:
RMgX + HA → RH + MgXA(格氏试剂被破坏)
因此:制备格氏试剂时分子中不能含上述基团;必须无水无氧操作。
炔基格氏试剂
端炔与格氏试剂的反应
端炔的C≡C−H具有弱酸性(pKa≈25),可被格氏试剂夺取活泼氢:
RC≡CH + R'MgX → RC≡CMgX + R'H
所得炔基格氏试剂 RC≡CMgX 是强亲核试剂,可与卤代烃发生SN2取代:
RC≡CMgX + R'X → RC≡CR' + MgX2
合成意义:这是合成非对称内炔烃(RC≡CR')的重要方法。要求R'X为伯卤代烃(SN2条件),叔卤代烃会发生消除。
格氏试剂在乙醚中不溶的情况:双键或苯环上的卤素(乙烯基/芳基卤)不易与Mg反应,需用THF(四氢呋喃)作溶剂,配位能力更强。
Corey-House 偶联
有机铜锂试剂
2RLi + CuI → R2CuLi(二烃基铜锂)
R2CuLi + R'X → R−R' + RCu + LiX
优势:交叉偶联效率高,选择性好(R'X最好是1°卤代烃)
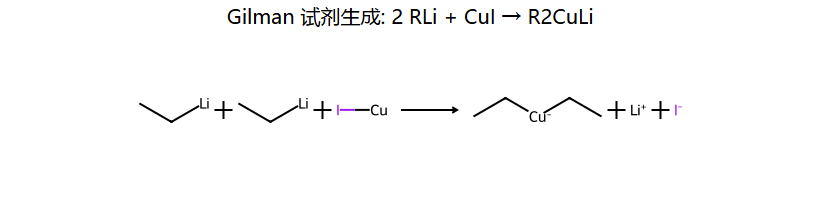
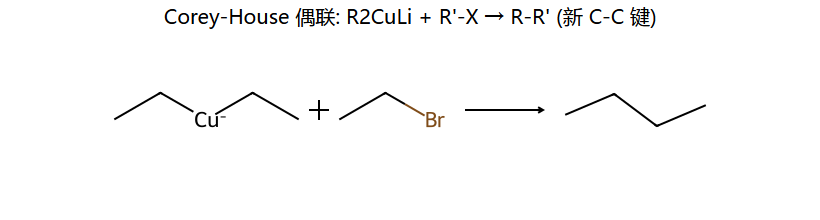
相比格氏试剂偶联(产率差),Gilman 试剂的 Cu 中心更软、选择性更好,能精准把 R 安到 R'X 的 C 上,新 C-C 键直接得到。
9.12 消除取向
Saytzeff 规则 vs Hofmann 规则
| Saytzeff(查依采夫) | Hofmann(霍夫曼) |
|---|
| 产物 | 双键上取代基最多的烯烃 | 双键上取代基最少的烯烃 |
| 条件 | 普通碱(NaOH, NaOEt) | 大位阻碱(t-BuOK, LDA) |
| 或 | 普通离去基团 | 大离去基团(如-NR3+) |
| 原因 | 热力学稳定性 | 位阻控制 → 碱只能夺最外面的H |
9.13 卤代烃的鉴别
AgNO3/醇溶液鉴别法
RX + AgNO3/醇 → AgX↓ + RONO2(SN1反应)
- 沉淀生成速度反映SN1活性:3° > 2° > 1°
- 烯丙基/苄基卤代烃:室温立即沉淀
- 乙烯基/芳基卤代烃:不反应
区分3-溴丙烯(烯丙型)、4-溴-2-烯(仲) 和 5-溴-2-烯(伯):
3-溴丙烯 → 室温快速沉淀;4-溴-2-烯 → 加热黄色沉淀;5-溴-2-烯 → 不反应或极慢
课后习题精选
1. 命名:写出分子式为 C
4H
9Br 的所有卤代烃结构式并命名。
答案
C4H9Br 含4个碳、9个氢、1个溴,不饱和度=0,为饱和卤代烷。
(1) CH3CH2CH2CH2Br — 1-溴丁烷(伯)
(2) CH3CH2CHBrCH3 — 2-溴丁烷(仲)
(3) (CH3)2CHCH2Br — 1-溴-2-甲基丙烷(伯)
(4) (CH3)3CBr — 2-溴-2-甲基丙烷(叔)
2. 自由基卤代:下列化合物哪些可由相应烃经光照单卤代制得?(1)CH
3CH
2Cl (2)CH
3CH
2CH
2CH
2Cl (3)(CH
3)
3CCH
2Cl (4)(CH
3)
2CHOCH
3 (5) 氯代环己烷 (6)H
2C=CHCHCH
2Cl
答案
对于饱和卤代烃,观察分子中所有H的化学等价性:若所有H化学等价,则能通过单卤代得到纯产物。对于不等价H的,产物为混合物。
(1) CH3CH2Cl:乙烷只有一种H环境 → 可以
(3) (CH3)3CCH2Cl:新戊烷中H有两种化学环境 → 混合物
(5) 氯代环己烷:环己烷所有H等价 → 可以
(6) 不饱和,不是简单自由基取代
答案:(1)(3)(5)(6)可由相应烃经自由基卤代制得(其中3和6产率有限)
3. 多步反应:完成下列转化:
(a) 环己烯 →(NaOH/EtOH)→ ? →(NBS)→ ? →(NaOH)→ ?
(b) 甲苯 →(Br
2/CCl
4)→ ? →(KOH/EtOH)→ ?
答案
(a) 环己烯不与NaOH/EtOH反应(不是卤代烃),保持为环己烯 → NBS进行烯丙位溴代 → 3-溴环己烯 → NaOH水溶液进行亲核取代(SN2) → 2-环己烯-1-醇
(b) 甲苯与Br2/CCl4:苯环不被催化(无Lewis酸),但甲苯侧链在非催化条件下也不反应(需要hv或NBS)。题目可能指Br2/hv → 苄基溴 C6H5CH2Br → KOH/EtOH消除 → 不可能(苄基溴无β-H?要看具体结构)。若底物为乙苯则:C6H5CHBrCH3 → KOH/EtOH → 苯乙烯
4. SN/E判断:2-溴丁烷分别与下列试剂反应,判断反应类型和主要产物:
(a) NaI/丙酮 (b) NaOH/H
2O (c) t-BuOK/t-BuOH (d) EtOH加热
答案
2-溴丁烷是仲卤代烃,四种反应都可能。
(a) NaI/丙酮:I−是强亲核弱碱,丙酮是极性非质子溶剂 → SN2 → 2-碘丁烷(构型翻转)
(b) NaOH/H2O:HO−既是亲核试剂又是碱,H2O是极性质子溶剂 → SN2 + E2竞争 → 2-丁醇 + 2-丁烯混合物
(c) t-BuOK/t-BuOH:强碱大位阻 → E2 → 2-丁烯(Saytzeff产物)+ 少量1-丁烯
注:t-BuOK虽是大碱偏Hofmann,但2-溴丁烷的Saytzeff产物位阻不大,仍以Saytzeff为主
(d) EtOH加热:弱亲核弱碱,极性质子溶剂,高温 → SN1 + E1 → 2-丁醇(外消旋) + 2-丁烯
5. 合成路线判断:下列合成是否正确?
(a) 烯烃 + HCl → RCl →(NaCN)→ RCN
(b) HO-苄基-Br →(Mg/乙醚)→ ? →(CO
2)→ ? →(H
+)→ COOH
答案
(a) 正确。烯烃加HCl得到氯代烃,然后CN−作为亲核试剂发生SN2取代(若是1°RCl)。但要注意过氧化物存在下HCl不发反Markovnikov加成。
(b) 错误。分子中含有−OH(活泼氢),格氏试剂不能与活泼氢共存!必须先保护OH基团(如转化为醚或酯),再制格氏试剂。
6. AgNO3鉴别:用简单化学方法区分以下化合物:
(1) 正氯丁烷、正溴丁烷、环己烯、对氯甲苯
答案
先用AgNO3/醇溶液:
• 正氯丁烷(1°)、正溴丁烷(1°):缓慢或加热生成沉淀
• 环己烯:不反应(不是卤代烃)
• 对氯甲苯(芳基氯):不反应
再用Br2/CCl4区分环己烯(褪色)和对氯甲苯(不褪色)。
区分正氯丁烷和正溴丁烷:沉淀颜色不同(AgCl白色,AgBr淡黄色)。
7. 转化:完成下列转化(每步写出试剂):
CH
3CH=CHCH
3 → (7步) → CH
3CH
2CH=CH
2
答案
2-丁烯 → 1-丁烯(双键位置异构化):
① 2-丁烯 + HBr → 2-溴丁烷(Markovnikov加成)
② 2-溴丁烷 + t-BuOK → 1-丁烯(Hofmann消除,大碱得取代少的烯)
或更直接:
① 2-丁烯 + HBr/ROOR → 不适用(只HBr有反Markovnikov)
实际上用大位阻碱E2消除2-溴丁烷的方案即可。
8. 立体化学:(R)-2-溴丁烷分别在以下条件下反应,画出产物立体化学:
(a) NaOH/DMSO (b) EtOH加热
答案
(a) NaOH/DMSO:SN2(强亲核试剂 + 极性非质子溶剂)→ 构型翻转 → (S)-2-丁醇
(b) EtOH加热:SN1(弱亲核、极性质子溶剂)→ 碳正离子中间体 → 外消旋化 → (R)-2-丁醇 和 (S)-2-丁醇 各约50%(外消旋体)
9. E2反式共平面:(1R,2R)-1-溴-1,2-二苯基丙烷进行E2消除,产物构型是E还是Z?
答案
E2要求H和Br处于反式共平面(anti)。画Newman投影式,将H和Br放在anti位置:
在anti构象中,两个苯基分别在同侧 → 消除后双键两侧的大基团在同侧 → 产物为(Z)-构型
这说明E2的立体化学是anti消除(反式消除),底物的立体构型决定了产物烯烃的E/Z构型。
10. 格氏试剂合成:从溴苯和不超过2个碳的有机物出发,合成3-苯基-1-丙醇。
答案
目标:C6H5CH2CH2CH2OH(比苯多3个碳的伯醇)
① C6H5Br + Mg/THF → C6H5MgBr(苯基格氏试剂)
② C6H5MgBr + 环氧乙烷 → C6H5CH2CH2OMgBr →(H3O+)→ C6H5CH2CH2OH(2-苯基乙醇,增2C)
这只增加了2个碳,得2-苯基乙醇。要得3-苯基-1-丙醇(增3C),需要:
① C6H5MgBr + CH2=CHCHO(丙烯醛,3C但超出2C限制)
或换思路:
① C6H5Br + Mg → C6H5MgBr
② + 环氧乙烷 → C6H5CH2CH2OH
③ + HBr → C6H5CH2CH2Br
④ + Mg → C6H5CH2CH2MgBr
⑤ + HCHO → C6H5CH2CH2CH2OH(3-苯基-1-丙醇)
11. SN1重排:3-甲基-2-丁醇与HBr反应产物是什么?会发生重排吗?
答案
(CH3)2CHCHOHCH3 + HBr →
醇先质子化:OH2+离去生成仲碳正离子 → 1,2-甲基迁移 → 叔碳正离子(CH3)3C+CH3 → Br−进攻
产物:2-溴-2-甲基丁烷(重排产物)+ 少量3-溴-2-甲基丁烷(未重排)
仲碳正离子 → 叔碳正离子(更稳定),所以会发生重排。
12. 综合:某化合物A分子式C
5H
10,不饱和度为1。A与HBr反应得B,B与KOH醇溶液消除得C,C与顺丁烯二酸酐Diels-Alder反应得D。写出A→D结构。
答案
A(C5H10,不饱和度1):可能是环戊烷或戊烯。
若A=环戊烯:
A(环戊烯) + HBr → B(溴代环戊烷) → KOH/醇消除 → C(环戊二烯?需要两次消除) → 环戊二烯与顺丁烯二酸酐D-A反应 → D(降冰片烯二酸酐)
实际上:A为环戊烯 → HBr → 3-溴环戊烯(烯丙位) → KOH/醇 → 环戊二烯(共轭二烯) → + 顺丁烯二酸酐 → 降冰片烯二酸酐(Diels-Alder产物)
历年真题原型(SJTU A 卷实证)
真题原型 1(A 卷 三-5,完成反应)——SN1 消旋 vs SN2 翻转对照
手性
2-氯戊烷 分别 ① AgNO
3/乙醇 ② NaI/丙酮,写产物立体化学。
完整解法步骤
① AgNO3/乙醇:Ag+ 拉走 Cl−、极性质子溶剂、2° 底物 → SN1 → 平面 sp2 碳正离子 → 硝酸根两面进攻 → 外消旋(R + S 各约 50%)的硝酸酯。
② NaI/丙酮(Finkelstein):I− 强亲核弱碱、极性非质子溶剂 → SN2 → 背面进攻 → Walden 翻转的 2-碘戊烷(构型字母翻转)。
对照核心:同一手性底物,条件一换立体结果就反过来——AgNO3/醇 = SN1 = 消旋;NaI/丙酮 = SN2 = 翻转。
真题原型 2(A 卷 二-9,选择/速率)——SN2 二级速率定量
CH
3I + OH
− 走 S
N2。若 [CH
3I] 变为原来 3 倍、[OH
−] 变为原来 1/2,反应速率如何变化?
完整解法步骤
SN2 二级速率方程:$v=k[\text{CH}_3\text{I}][\text{OH}^-]$。
两个浓度都要代入:$v' = k\cdot(3[\text{CH}_3\text{I}])\cdot(\tfrac12[\text{OH}^-]) = 3\times\tfrac12\,v = 1.5\,v$。
→ 速率提高 50%。
易错:只乘 RX 那个浓度(漏掉 Nu)就会答错;SN1 才是 $v=k[\text{RX}]$、与 [Nu] 无关。
真题原型 3(A 卷 六-2,合成)——Grignard + CO2 增 1 碳
由 (CH
3)
3COH 出发合成
2,2-二甲基丙酸(增 1 个碳)。
完整解法步骤
叔醇不能直接做格氏(也无 α-H 可直接氧化增碳),必须走卤代→格氏→CO2:
① (CH3)3COH + HBr → (CH3)3CBr(叔醇极易 SN1 卤代)
② (CH3)3CBr + Mg/无水乙醚 → (CH3)3CMgBr
③ + CO2(干冰)→ (CH3)3CCOO−MgBr+
④ + H+/H2O → (CH3)3CCOOH(2,2-二甲基丙酸 / 新戊酸)
增碳速记:RMgX + CO2 → 羧酸(+1C);+ 环氧乙烷 → 伯醇(+2C);+ 醛 → 仲醇;+ 酮 → 叔醇;+ D2O → C–D。
📌 必背块(机理判据·速率·活性序)
SN1 / SN2 / E1 / E2 速率与立体(默写线)
| SN2 | SN1 | E2 | E1 |
|---|
| 速率 | $v=k[\text{RX}][\text{Nu}]$ | $v=k[\text{RX}]$ | $v=k[\text{RX}][\text{B}]$ | $v=k[\text{RX}]$ |
| 底物 | CH3>1°>2° | 3°>2° | 3°>2°>1° | 3°>2° |
| 立体 | Walden 翻转 | 外消旋 | 反式共平面 | — |
| 重排 | 无 | 有 | 无 | 有 |
活性序 / 离去能力 / 亲核性
SN 活性:烯丙基≈苄基 >> 3°(SN1) / CH3>1°(SN2) >> 乙烯基≈芳基≈桥头碳(不反应)。
离去能力:–OTs≈–OMs > I− > Br− > Cl− > F−(共轭酸越强越好离去)。
亲核性(质子溶剂同族):I− > Br− > Cl− > F−;负离子 > 中性(HO−>H2O)。
消除取向 + 溶剂口诀
普通碱(NaOH/NaOEt) → Saytzeff(多取代烯);大位阻碱(t-BuOK)/大离去基(–NR3+) → Hofmann(少取代烯)。
醇中消除、水中代:KOH/醇 → 消除为主;KOH/水 → 取代为主。极性非质子(DMSO/丙酮)利 SN2;极性质子(H2O/ROH)利 SN1。
Grignard 三忌 + 增碳表
三忌:① 不能有活泼 H(–OH/–NH/–SH/–COOH/–C≡CH,会自杀失活)② 必须无水无氧 ③ 乙烯基/芳基卤用 THF 不用乙醚。
增碳:+CO2→羧酸(+1C);+环氧乙烷→伯醇(+2C);+HCHO→伯醇(+1C);+RCHO→仲醇;+R2CO→叔醇;+D2O→C–D。
⚠️ 易错点
1. 速率题两个浓度都要乘:SN2 是 $v=k[\text{RX}][\text{Nu}]$,改了底物又改了亲核试剂浓度时两项都要代入(如 3×½=1.5)。SN1 与 [Nu] 无关。
2. 碱性 ≠ 亲核性:I− 碱性弱但亲核性强(大原子可极化)→ 偏 SN2;t-BuO− 碱性强但亲核性差(位阻大)→ 偏 E2。要纯取代避消除时用弱碱强亲核体(CH3COO−/I−/N3−)。
3. E2 必须反式共平面:环己烷上 H 和 X 要处于 1,2-双直键(diaxial) 才能消除;只有一个 β-H 满足 anti 时即使不是 Saytzeff 产物也只能得它。底物构型决定产物 E/Z。
🔴 你在这卡过(FXY 个人薄弱点 → 怎么破)
卡点 A:碳正离子重排(新戊基 / Wagner-Meerwein 1,2-迁移)
你卡在哪:新戊醇 (CH3)3C–CH2OH + HCl/ZnCl2 为什么产物骨架变成带乙基的 2-氯-2-甲基丁烷?(同一题两周内问了 2 次,系统连发重复弱点预警)本章的"新戊基溴 SN1 重排"是同一个机理。
怎么破(4 步默写):
① 走 SN1:脱去离去基 → 极不稳的 1° 碳正离子 (CH3)3C–CH2+。
② 旁边叔碳上一个 CH3 带着电子对 1,2-迁移 过来。
③ 变成稳定的 3° 碳正离子 (CH3)2C+–CH2CH3(骨架已带乙基!)。
④ Nu−(Cl−)捕获 → 2-氯-2-甲基丁烷。
判别钉子 + 避坑:
• HX/ZnCl2 / 浓 H2SO4 / 醇脱水加热 + 产物骨架对不上 = 一定重排。
• 别套 SN2 直接写 OH→Cl(那会错成 (CH3)3C–CH2Cl)。
• 合成里要避开重排就用 SOCl2 / PBr3(走 SN2、不经碳正离子);HX/ZnCl2 必重排。
• 迁移方向:β-C 上有 H 迁 H、无 H 有 CH3 迁 CH3,原则是迁完碳正离子级数升高。
卡点 B:醇→碘代物两步法(OTs 活化 + SN2 Walden 翻转)
你卡在哪:C2H5–CHD–OH 经 TsCl/吡啶 → NaI/丙酮 看不懂(同一题问了 2 次)。本章 9.10 制备里的 "醇→OTs→I" 就是它。
怎么破:OH 太顽固,I− 直接踢不动 →
① TsCl/吡啶 把 OH 活化成超级好离去的 –OTs;
② NaI/丙酮 做 SN2,I− 背面进攻把 OTs 换成 I → 构型翻转一次(Walden)。
避坑:丙酮是溶剂不是反应物;D(氘)只是同位素标记,化学性质同 H,全程不参与、不迁移,别被它干扰判断手性中心。
考前提醒
SN1/SN2/E1/E2判断是必考题!三步法:看底物(1°/2°/3°) → 看试剂(亲核性/碱性/位阻) → 看溶剂(质子/非质子)
SN2构型翻转,SN1外消旋——立体化学是常考点
E2反式共平面——环己烷上H和X必须1,2-双直键(diaxial),这决定了产物构型
格氏试剂三大忌:① 不能有活泼氢(OH/NH/SH/COOH/C≡CH) ② 必须无水无氧 ③ 乙烯基/芳基卤用THF不用乙醚
活性顺序:烯丙基≈苄基 >> 叔 > 仲 > 伯 >> 乙烯基≈芳基≈桥头碳(不反应)
Saytzeff vs Hofmann:普通碱→Saytzeff(取代多);大碱(t-BuOK)→Hofmann(取代少)
碱性≠亲核性:I−碱性弱但亲核性强(SN2);t-BuO−碱性强但亲核性差(E2)
极性非质子溶剂(DMSO/DMF)利SN2;极性质子溶剂(H2O/ROH)利SN1