核心:第一步生成碳正离子是决速步,碳正离子的稳定性决定了区域选择性和立体选择性!
真分子上的亲电加成 2 步 (丙烯 + HBr)
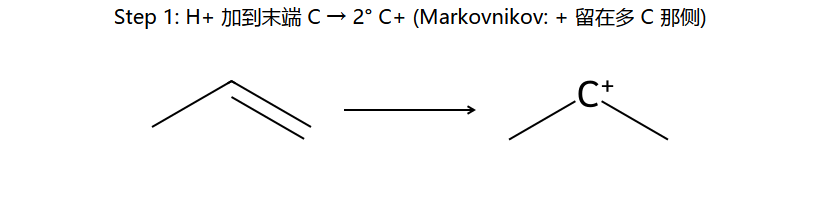
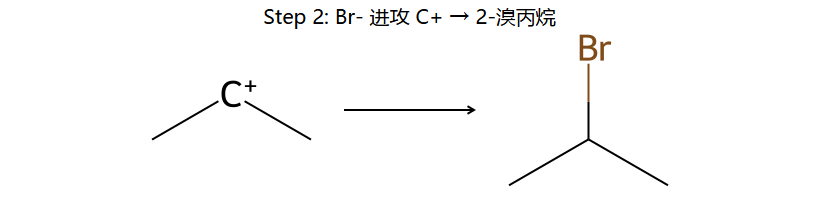
看图重点:H+ 只能加到末端 CH2 那个 C,因为这样正电才会留在中间的 C 上(2° 稳)。反过来加,正电就在末端 1° 上(不稳)。区域选择性的本质就是谁能给出更稳定 C+。
①原子序数大者优先 ②第一个原子相同则比较下一个 ③双键按两个单键计算
Z(zusammen):两个优先基团在同侧;E(entgegen):在异侧
氢化热越小 → 烯烃越稳定。取代基越多 → 超共轭效应越强 → 越稳定
稳定性顺序:四取代 > 三取代 > 二取代(反式>顺式) > 一取代 > 无取代
①沸点随分子量增大而升高;低级烯烃(C₂~C₄)为气态,C₅以上为液态
②Z-异构体沸点略高于E-异构体:Z式偶极矩较大,分子间作用力更强
③难溶于水,易溶于有机溶剂(非极性分子,"相似相溶")
电负性:Csp > Csp² > Csp³
原因:s成分越多,电子越靠近原子核,对电子束缚力越强(表现为电负性越大)
应用:乙炔C-H的酸性 > 乙烯C-H > 乙烷C-H
看图重点:H+ 只能加到末端 CH2 那个 C,因为这样正电才会留在中间的 C 上(2° 稳)。反过来加,正电就在末端 1° 上(不稳)。区域选择性的本质就是谁能给出更稳定 C+。
不对称烯烃加HX:H加到含H较多的双键碳 → 生成更稳定的碳正离子
本质:经过更稳定的碳正离子中间体(能量更低的过渡态)
碳正离子稳定性:
苄基 ≈ 烯丙基 > 3° > 2° > 1° > CH₃⁺ > 乙烯基
稳定化因素:超共轭(烷基)、共振(苄基/烯丙基)、杂原子孤对电子
①1,2-H迁移:H从邻碳带着电子对迁移到正电荷碳
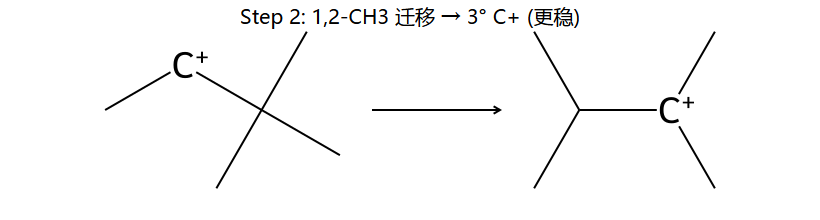
②1,2-甲基迁移:甲基(或其他烷基)带电子对迁移
③方向:总是从不稳定→更稳定碳正离子
④环扩张:四/五元环→五/六元环(张力释放)
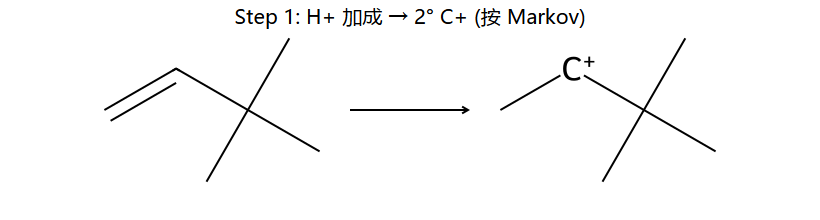
①H⁺加到C1(马氏规则) → 在C2形成2°碳正离子
②C3上甲基1,2-迁移到C2 → 变为3°碳正离子
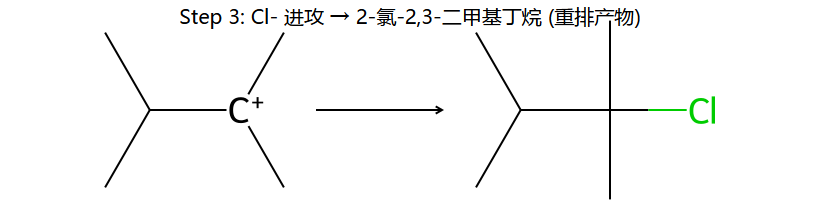
③Cl⁻进攻3°碳 → 产物为2-氯-2,3-二甲基丁烷
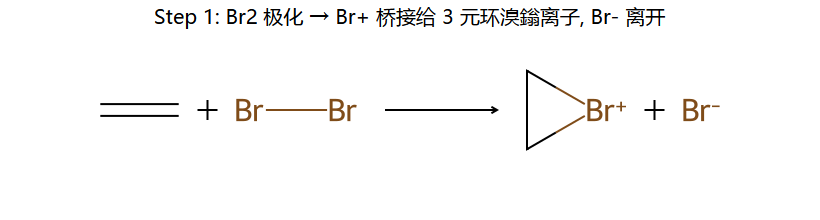
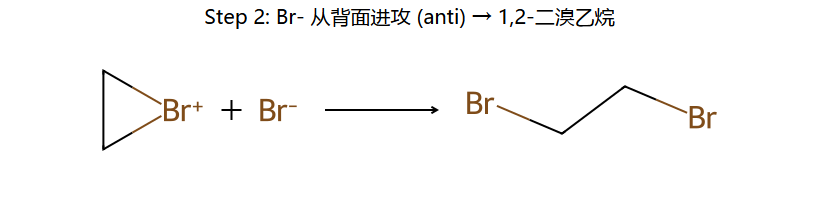
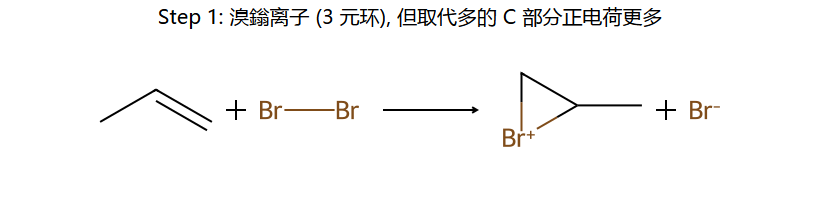
①Br₂被π电子极化 → Br⁺形成三元环桥接(溴鎓离子)
②Br⁻只能从三元环背面(anti)进攻 → 两个Br在双键两侧
③环己烯加Br₂ → 反式-1,2-二溴环己烷(两个Br为trans-diaxial)
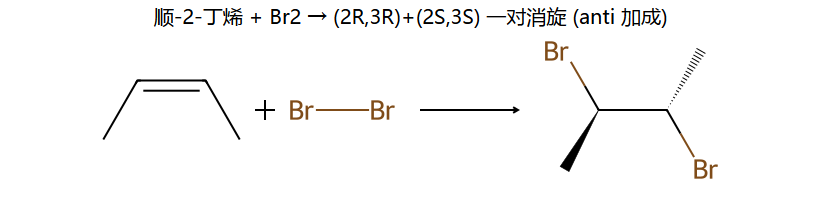
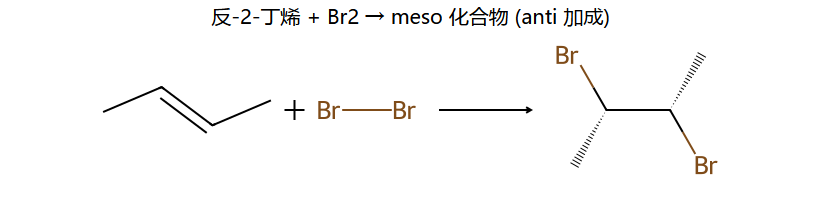
看图重点:anti 加成的立体后果——同一根 C=C,顺式底物 vs 反式底物给出不同的立体异构体(一个消旋对 vs 一个 meso)。这就是为什么烯烃加 Br2 题里"顺反"必须看清楚。
Cl原子较小,不能有效形成三元环 → 经开放碳正离子中间体
结果:Cl₂加成的立体选择性不如Br₂严格,可能得到顺式+反式混合物
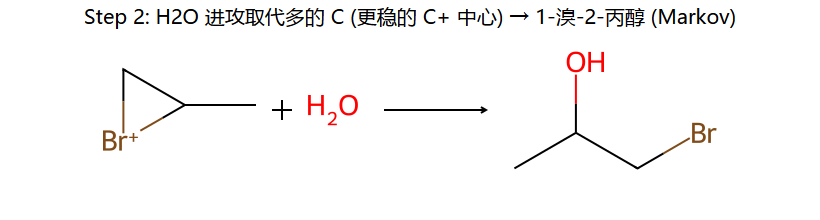
烯烃 + Br₂/H₂O → 溴醇(halohydrin)
机理:先形成溴鎓离子 → H₂O(而非Br⁻)作亲核试剂进攻
区域:OH加到取代多的碳(马氏),Br加到取代少的碳
立体:仍为反式加成(OH和Br在两侧)
烯烃 + H₂SO₄(浓) → 硫酸氢酯(ROSO₃H) → 水解 → 醇 + H₂SO₄
总结果:马氏加水(H加到含H多的碳,OH加到含H少的碳)
烯烃活性与硫酸浓度的关系:烷基取代越多 → 形成碳正离子越稳定 → 所需H₂SO₄浓度越低
①烯烃 + 醇(H⁺催化) → 醚(马氏方向,碳正离子被ROH捕获)
②烯烃 + 羧酸(H⁺催化) → 酯(碳正离子被RCOOH捕获)
机理与HX加成类似:H⁺先加到双键形成碳正离子 → 亲核试剂(ROH或RCOOH)进攻碳正离子
F₂ > Cl₂ > Br₂ > I₂
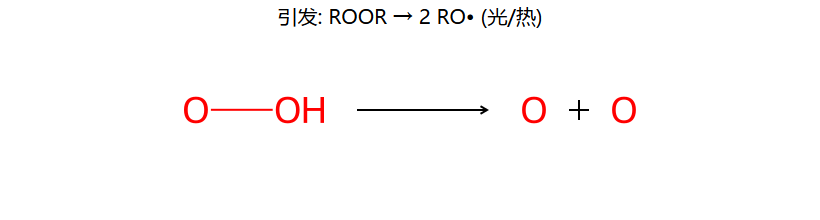
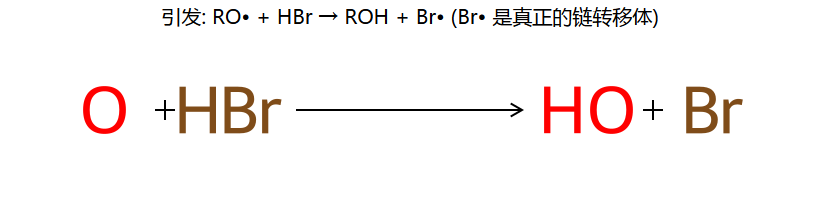
①引发:ROOR → 2RO· → RO· + HBr → ROH + Br·
②增长:Br·加到含H多的碳(因为生成更稳定自由基) → 碳自由基再夺HBr的H
③终止:自由基偶合
结果:反马氏加成 — Br加到含H多的碳,H加到含H少的碳
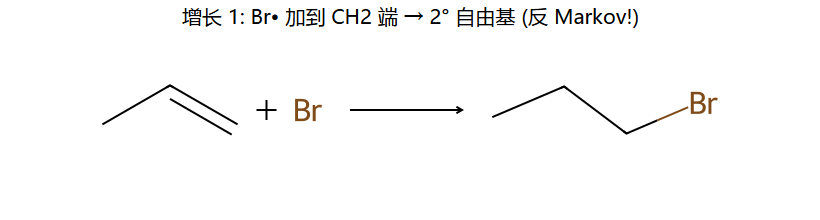
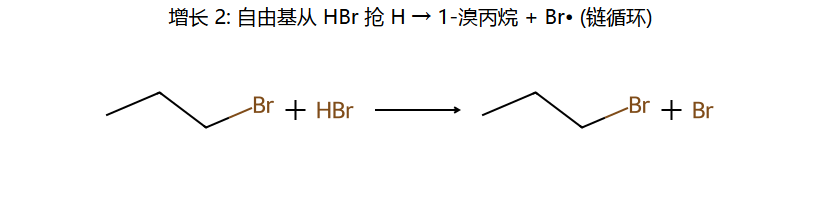
反 Markov 的根源:Br• 是中性的,没有"正电要找稳定地方"的压力。Br• 先加到烯烃,加在哪一端由剩下的自由基稳不稳决定。Br 加在末端 CH2 → 剩下 2° 自由基(更稳)→ Br 落到 1° 那端,与"Markov 给出的 Br 落到 2°"恰好相反。
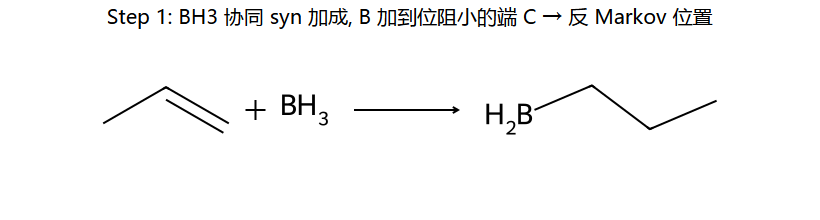
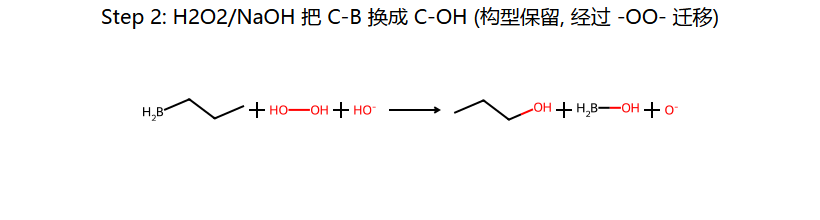
①反马氏:OH加到含H多的碳(B先加到位阻小的碳)
②顺式加成(syn):H和OH加到双键同侧(四元环过渡态协同反应)
③无重排:不经过碳正离子中间体
| 试剂/条件 | 产物 | 立体化学 | 特点 |
|---|---|---|---|
| KMnO₄ 稀冷/中性 | 邻二醇(cis) | 顺式加成 | 双键不断裂;可做检验 |
| OsO₄ + NMO | 邻二醇(cis) | 顺式加成 | 催化量OsO₄;选择性好 |
| KMnO₄ 浓热/酸性 | 羧酸 + 酮 | — | 双键断裂氧化 |
| O₃ → Zn/H₂O(还原) | 醛 + 酮 | — | 臭氧化-还原裂解 |
| O₃ → H₂O₂(氧化) | 羧酸 + 酮 | — | 臭氧化-氧化裂解 |
| mCPBA(过氧酸) | 环氧化物 | 顺式加成 | 构型保持;协同机理 |
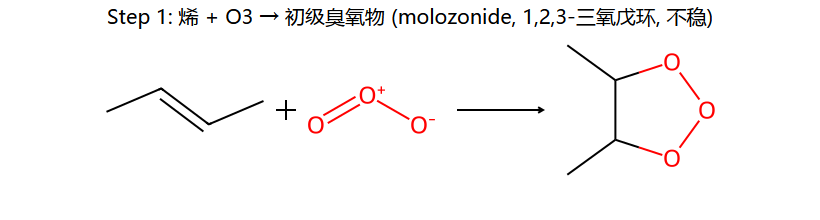
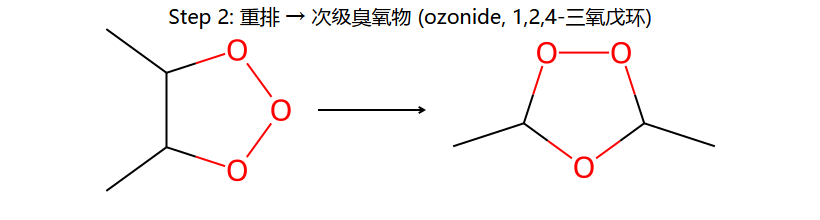
C=C + O₃ → 臭氧化物(molozonide) → 重排为ozonide → 还原裂解得醛/酮
应用:已知产物(醛/酮)可反推原来的烯烃结构(两个C=O拼回C=C)
将两个羰基碳用双键连接:(CH₃)₂C=CH₂ → 2-甲基丙烯(异丁烯)
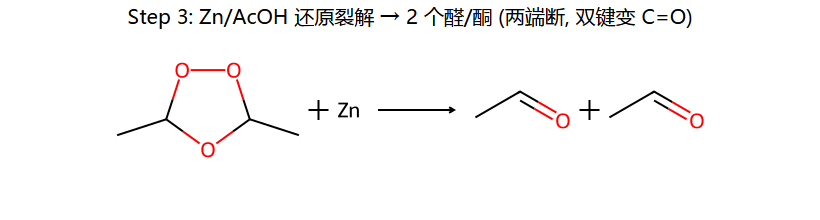
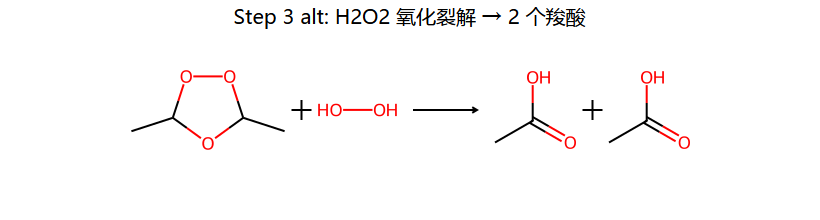
还原 vs 氧化裂解:还原条件 (Zn/AcOH 或 Me2S) 给醛/酮;氧化条件 (H2O2) 把 R-CHO 进一步氧化成 R-COOH,但叔位 C 上没 H,所以酮还是酮。
特点:协同机理(一步完成),顺式加成,双键上原来的顺反构型完全保持
富电子烯烃反应更快(取代越多越快)
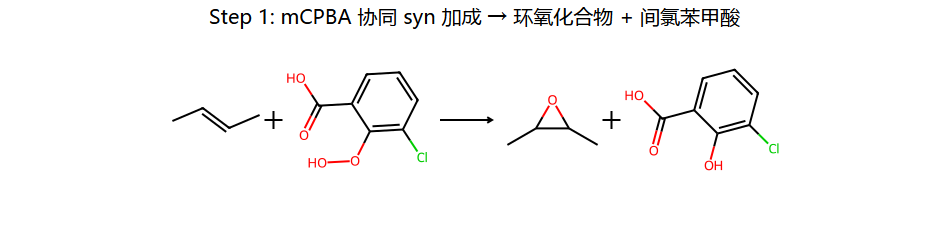
蝴蝶过渡态:mCPBA 的 O-O 单键和 O-H 之间的协同重排,让一个氧整体从 mCPBA 跳到烯烃 → 三元环 → 环氧产物。整个过程没有中间体(一步),所以双键的顺反构型完全保持。
H₂/Pd(或Pt、Ni) → 顺式加成(两个H从金属表面同侧加入)
氢化热比较 → 取代越多的烯烃越稳定 → 氢化越慢(放热越少)
烯丙基自由基有p-π共轭稳定化(电子离域到相邻π体系)
NBS的作用:维持低浓度Br₂ → 有利于取代(自由基链反应)而非加成
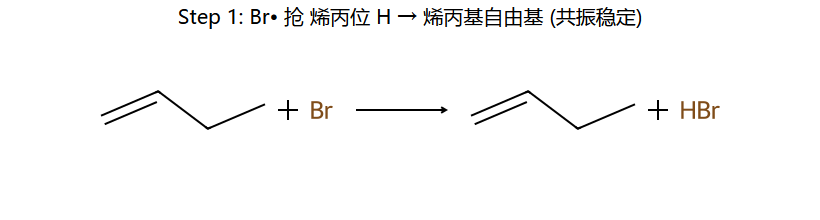
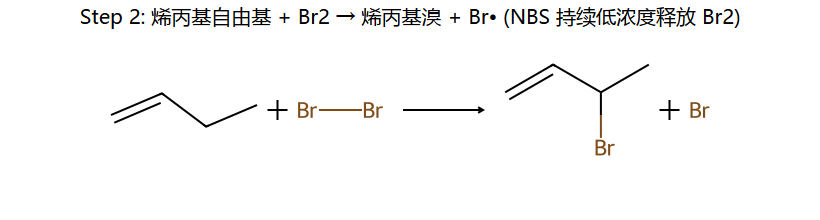
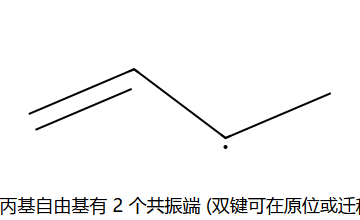
关键:烯丙基自由基有 p-π 共轭 稳定化 → 自由基 + Br• 抢的是 烯丙位 H(不是末端 H 或中间 sp2 H)。NBS 不直接放 Br•,而是慢释放 Br2,让浓度保持很低 → 自由基取代占主导,加成被抑制。
Grubbs催化剂:两个烯烃交换双键两侧的基团
R¹CH=CHR² + R³CH=CHR⁴ → R¹CH=CHR³ + R²CH=CHR⁴
n CH₂=CH₂ → [-CH₂-CH₂-]n(聚乙烯)
①自由基聚合(高压法):
②Ziegler-Natta催化聚合(低压法):
ROH → (H₂SO₄, 170°C) → 烯烃 + H₂O
机理:①醇质子化 → ②失水生成碳正离子 → ③消除β-H得烯烃
Zaitsev(扎依采夫)消除规则:消除反应优先生成取代最多(最稳定)的烯烃
碳正离子中间体可从C1或C3失去H:
失去C3的H → 2-丁烯(更稳定,双取代烯烃)= 主产物(Zaitsev规则)
失去C1的H → 1-丁烯(单取代烯烃)= 次产物
醇脱水活性:3°醇 > 2°醇 > 1°醇(碳正离子越稳定越容易形成)
RX + KOH/EtOH(强碱/醇溶液) → 烯烃 + KX + H₂O
条件:强碱(KOH、NaOEt等) + 醇溶剂(促进消除而非取代)
区域选择性:遵循Zaitsev规则 → 优先生成取代最多的烯烃
立体要求:E2为反式共平面消除 → H和X必须处于anti-periplanar构象
R(X)CH-CH(X)R' + Zn → 烯烃 + ZnX₂
试剂:锌粉/乙醇 或 NaI/丙酮
特点:条件温和,不经过碳正离子(不重排),可保留原有双键位置
应用:常用于炔烃加卤→邻二卤代物→脱卤得烯烃的合成路线中保护/去保护双键
| 反应 | 区域选择性 | 立体选择性 | 中间体 |
|---|---|---|---|
| HX加成 | 马氏(Markovnikov) | 无特定立体 | 碳正离子(可重排) |
| H₂O/H⁺加成 | 马氏 | 无特定立体 | 碳正离子(可重排) |
| Br₂加成 | — | 反式(anti) | 溴鎓离子 |
| Br₂/H₂O | OH在多取代碳 | 反式(anti) | 溴鎓离子 |
| HBr/ROOR | 反马氏 | 无特定立体 | 自由基 |
| BH₃→H₂O₂ | 反马氏 | 顺式(syn) | 四元环过渡态 |
| H₂/Pt | — | 顺式(syn) | 表面吸附 |
| OsO₄(KMnO₄稀) | — | 顺式(syn) | 环酯中间体 |
| mCPBA环氧化 | — | 顺式(syn) | 协同(蝶形) |
考点:过氧化物效应(仅 HBr 有效)+ 孤立二烯的区域选择性。
第①步:HBr + ROOR → 自由基机理,由 Br· 引发。
第②步:Br· 优先加到能生成更稳定自由基的那根双键 = 三取代双键 (CH₃)₂C=CH−(端基乙烯加成只能给 1° 自由基,不利)。
第③步:反马 —— Br 加到取代少的内侧碳、H 加到 (CH₃)₂C 那个取代多的碳;端基乙烯不动。
产物:(CH₃)₂CH−CHBr−CH₂−CH=CH₂。
⚠️ 这是孤立 1,4-二烯,反马是普通 1,2-加成(Br 不在端碳、也不是 1,4!)。只有共轭二烯才考虑 1,4。
考点:加氢快慢与烯烃稳定性反向。
判法:取代基越少 → 烯烃越不稳定(氢化热越大、放热越多)→ 越易吸附到金属表面 → 加氢越快。故答案 = CH₂=CH₂(乙烯,无取代)(B)。
反直觉点:四取代烯烃最稳定,但因位阻大、能量低,加氢最慢。别把"稳定"和"反应快"画等号。
HX / H₂O·H⁺ → 马氏,经碳正离子,会重排,无特定立体;
HBr/ROOR → 反马(仅 HBr!自由基),无特定立体;
Br₂/CCl₄ → 经溴鎓离子,反式(anti)加成;Br₂/H₂O → OH 加多取代碳(马氏向)、Br 加少取代碳,仍反式;
BH₃→H₂O₂/OH⁻(硼氢化氧化)→ 反马 + 顺式(syn),不经碳正离子、不重排。
稀冷/中性 KMnO₄ → 顺式邻二醇(双键不断,可做检验);浓热/酸性 KMnO₄ 或 O₃→H₂O₂ → 断键(R₂C= → 酮,RCH= → 酸);O₃→Zn/H₂O → 醛+酮;mCPBA → 环氧(顺式,构型保持)。
H₂/Pt(Pd,Ni) → 顺式加成;加氢速率 ∝ 烯烃不稳定度(取代越少越快)。NBS/hν → 烯丙位自由基溴代(取代不加成,经烯丙基自由基 p-π 共振)。
碳正离子:苄基 ≈ 烯丙基 > 3° > 2° > 1° > CH₃⁺ > 乙烯基。烯烃稳定性:四取代 > 三取代 > 二取代(反>顺) > 一取代 > 乙烯。
命名步骤:①找含双键的最长碳链 ②从靠近双键的一端开始编号 ③标出取代基位置和名称 ④用CIP规则判断E/Z
例:CH₃CH=C(CH₃)₂ → 2-甲基-2-丁烯(三取代烯烃,无E/Z,因为一端两个相同基团)
排序依据:①共振稳定(苄基/烯丙基) ②取代度(3°>2°>1°) ③超共轭
苄基碳正离子 > 烯丙基碳正离子 > 叔碳正离子 > 仲碳正离子 > 伯碳正离子 > 甲基正离子
苄基特别稳定因为正电荷可以离域到整个苯环(4个共振式)
分析方法:
①确定加成方式(syn/anti) ②画出产物立体结构 ③判断是否有对称面
情况1:对称烯烃(如2-丁烯) + Br₂ → 反式加成
(E)-2-丁烯 + Br₂ → 内消旋体(meso) → 无旋光性
(Z)-2-丁烯 + Br₂ → (R,R)+(S,S)外消旋体 → 无旋光性(混合物)
情况2:不对称烯烃 → 通常得外消旋体(无旋光性)
结论:加成反应一般不产生旋光性,除非使用手性试剂/催化剂
(a) 1-甲基环己烯 + HBr → 1-溴-1-甲基环己烷(马氏,3°碳正离子)
(b) 1-甲基环己烯 + HBr/ROOR → 1-甲基-2-溴环己烷(反马氏,自由基)
(c) 1-甲基环己烯 + Br₂ → trans-1,2-二溴-1-甲基环己烷(反式加成)
(d) 1-甲基环己烯 + BH₃→H₂O₂ → trans-2-甲基环己醇(反马氏+顺式加成)
3,3-二甲基-1-丁烯 + HCl:
①H⁺加到C1 → C2形成2°碳正离子: (CH₃)₃C-CH⁺-CH₃
②C3上的甲基(或C3-C2的σ键)进行1,2-迁移 → C2变为3°碳正离子
③Cl⁻进攻 → 2-氯-2,3-二甲基丁烷
环扩张实例:甲基环丁烯+HCl → 初始形成环丁基碳正离子(张力大) → C-C键迁移扩张为环戊基碳正离子(张力释放) → 产物为氯代环戊烷衍生物
①Br₂/CCl₄:烯烃使溴褪色(加成),烷烃不反应
②稀冷KMnO₄:烯烃使紫色褪色(氧化为邻二醇),烷烃不反应
③浓H₂SO₄:烯烃溶解(质子化形成碳正离子),烷烃不溶
区分不同烯烃:催化加氢后鉴定产物;臭氧化后鉴定醛酮产物
环己烯 + Br₂ → trans-1,2-二溴环己烷
步骤1:Br₂靠近电子云 → π电子进攻Br-Br → 形成三元环溴鎓离子 + Br⁻
步骤2:Br⁻从溴鎓离子背面(anti)进攻 → SN2型开环
立体结果:两个Br必须处于环的trans位置(diaxial构象)
光学活性:环己烯是对称分子 → 加成得到的trans产物为内消旋体(有对称面) → 无旋光性
天然萜类(如薄荷醇、樟脑)的生物合成涉及:
①焦磷酸酯离去 → 形成烯丙基碳正离子
②碳正离子进攻分子内双键 → 新C-C键(环化)
③连续1,2-迁移/环扩张 → 最终产物骨架
④消除H⁺或被亲核试剂捕获 → 终止
这体现了碳正离子化学在自然界中的重要性!
异丁烯在H₂SO₄催化下的二聚反应:
①质子化:(CH₃)₂C=CH₂ + H⁺ → (CH₃)₃C⁺(叔碳正离子)
②碳正离子进攻另一分子异丁烯的双键:(CH₃)₃C⁺ + (CH₃)₂C=CH₂ → (CH₃)₃C-CH₂-C⁺(CH₃)₂(新的叔碳正离子)
③Zaitsev消除(失去β-H):生成取代最多的烯烃 → 2,4,4-三甲基-2-戊烯(主)或2,4,4-三甲基-1-戊烯(次)
关键:整个过程体现了碳正离子的生成→亲电加成→消除的串联反应